In 1953, James Watson, Francis Crick, and Maurice Wilkins unraveled the structure of deoxyribonucleic acid (DNA), earning them the Nobel Prize in Physiology/Medicine. This breakthrough provided the key to understanding the language of life, where the linear arrangement of four bases – adenine, thymine, guanine, and cytosine – held the secrets of orchestrating living activities. The code within the DNA, represented by sequences of codons, directs the synthesis of proteins, governing what, where, when, and how much protein is produced. The discovery of the DNA structure and the genetic code laid the foundation for a transformative journey – the quest to decipher the meaning of DNA sequences.
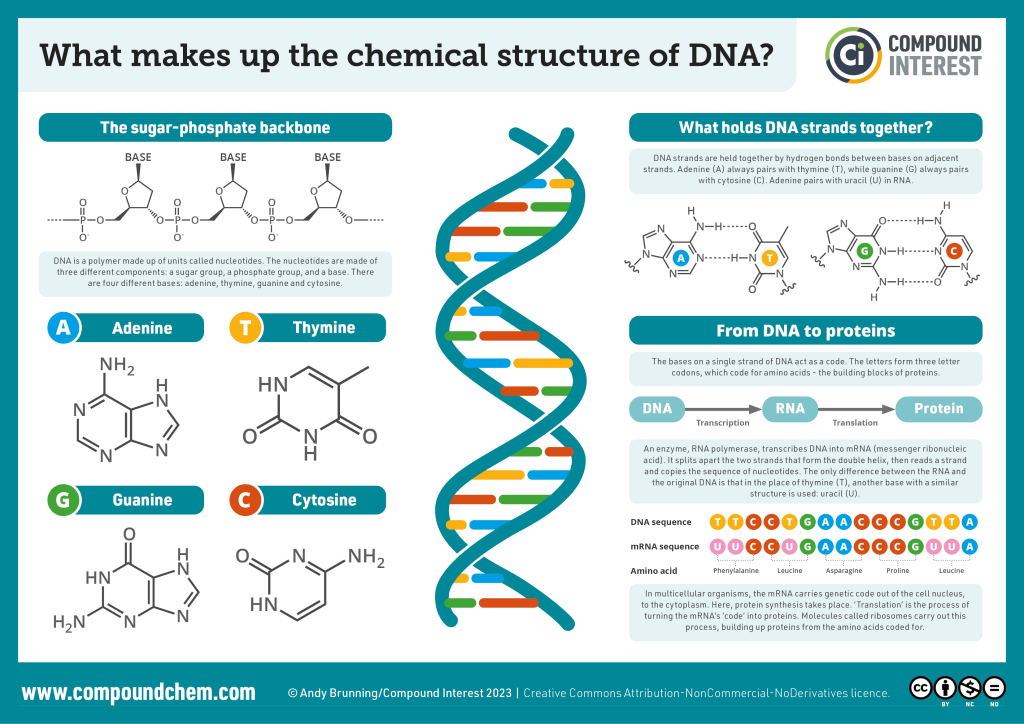
Purine and Pyrimidine Bases: The Foundation of Genetic Information
Purines. The basic purine structure comprises nine atoms, consisting of a six-membered pyrimidine ring fused with a five-membered imidazole ring. Within this structure, four nitrogen atoms are located at positions 1, 3, 7, and 9. The numbering of purine starts from the first nitrogen of the six-membered ring and proceeds counterclockwise, while the imidazole ring is numbered in a clockwise direction.
Purine bases form nucleosides by attaching to the 1’ carbon of pentoses through the ninth nitrogen atom. Notably, the purine ring’s electrons exhibit significant delocalization, providing it with unique electronic properties. Specifically, positions 3 and 7 within the purine ring possess an electron-rich nature, making them susceptible to electrophilic attacks. Conversely, positions 2, 6, and 8 are prone to nucleophilic attacks due to their electronic configuration. Within the context of nucleic acids, adenine and guanine are the two primary purine bases – adenine and guanine.
Adenine, characterized by its double-ring structure, forms hydrogen bonds with thymine in DNA (uracil in RNA) through complementary base pairing. This specific binding pattern underpins the remarkable fidelity of DNA replication, where one DNA strand serves as a template to synthesize its complementary strand. This purine-pyrimidine pairing establishes the basis for the well-known double helix structure of DNA. Guanine, structurally similar to adenine, also engages in hydrogen bonding with cytosine in DNA and RNA. The formation of three hydrogen bonds between guanine and cytosine further reinforces the stability and integrity of the DNA double helix. The purine-pyrimidine interactions play a crucial role in DNA repair, recombination, and other vital cellular processes.
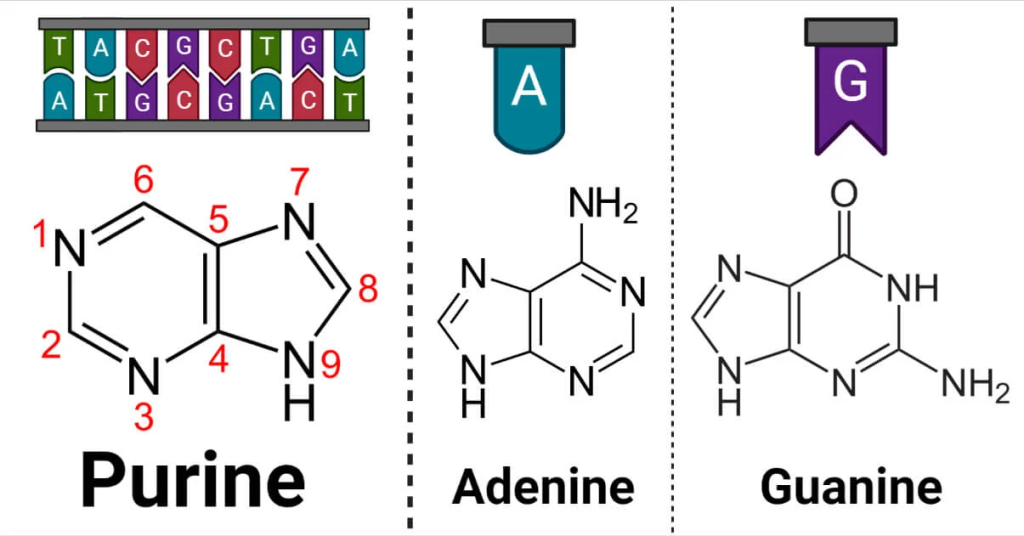
Pyrimidines. Pyrimidine bases, in contrast, are monocyclic aromatic compounds, featuring a single six-membered ring. Within this ring, four carbon atoms are linked to hydrogen or side chains, while two nitrogen atoms are situated at positions -1 and -3. The arrangement of carbon (C) and nitrogen (N) atoms in the pyrimidine ring involves a combination of single and double bonds, creating a dynamic and versatile structure. The unique arrangement of nitrogen (N) and carbon (C) atoms allows pyrimidine to bond with hydrogen or other functional groups, leading to the formation of various pyrimidine derivatives. These derivatives exhibit diverse properties and functions, contributing to a wide range of biological processes and chemical reactions. Three essential pyrimidines are cytosine, thymine, and uracil.
Cytosine, like guanine, engages in three hydrogen bonds to form base pairs with guanine. The precise arrangement of these base pairs allows for efficient and accurate genetic replication and gene expression. Thymine is unique to DNA, where it pairs with adenine through two hydrogen bonds. The adenine-thymine pairing provides a mechanism for the faithful transfer of genetic information from one generation to the next. Uracil, on the other hand, is found exclusively in RNA, replacing thymine. In RNA, uracil pairs with adenine during transcription, allowing RNA to serve as a transient messenger between DNA and protein synthesis machinery.
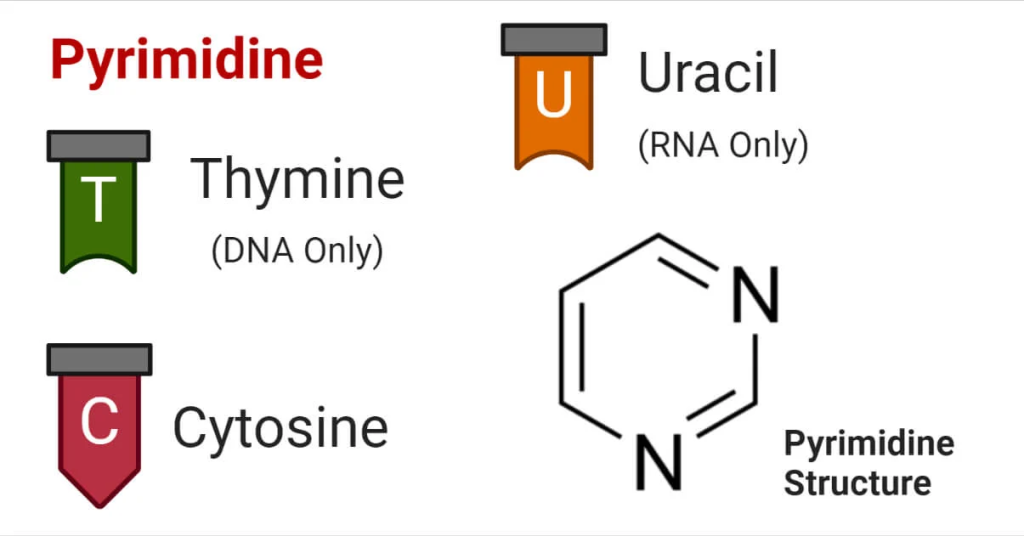
The complementary base pairing between purine and pyrimidine bases within DNA and RNA is a cornerstone of genetics and molecular biology. This precise arrangement ensures the accurate transmission of genetic information during replication and transcription processes. It also facilitates various cellular functions, such as DNA repair, recombination, and regulatory processes that govern gene expression. The structural properties of purine and pyrimidine bases contribute to the stability of the DNA double helix and RNA secondary structures. Understanding their interactions and roles in nucleic acids is essential for deciphering the molecular mechanisms that underlie life’s processes.
The First Revolution in DNA Sequencing
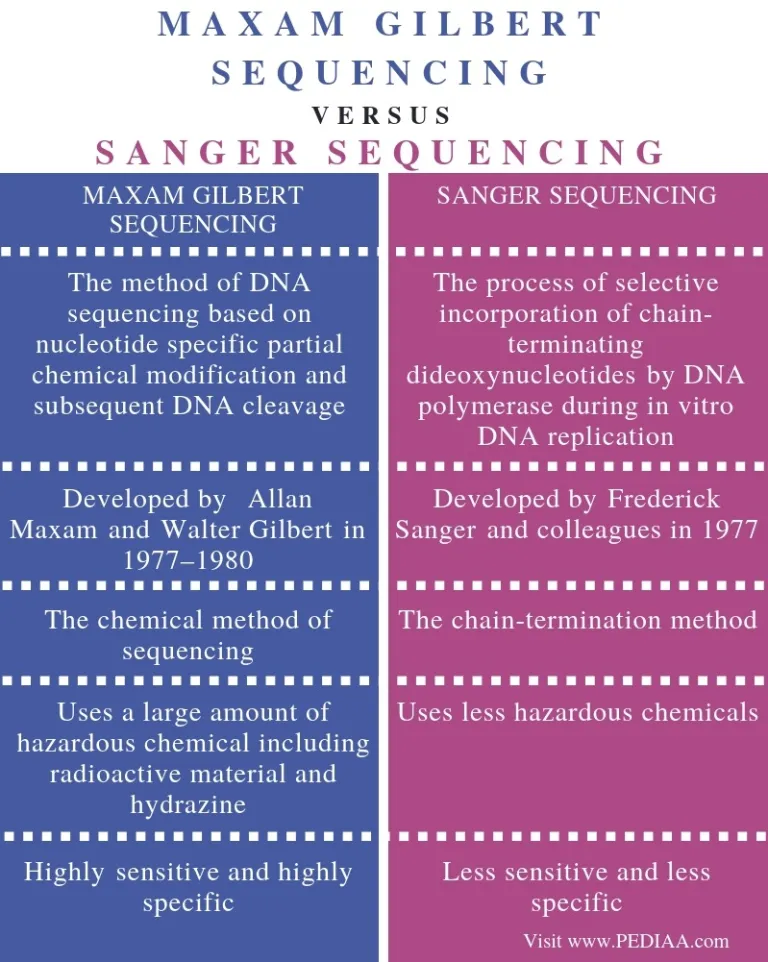
Sequencing DNA is essential for understanding its significance, and the race to develop better tools for this purpose has been intense. In the 1970s, two major competitors emerged: the Sanger sequencing method and the Maxam–Gilbert sequencing method. However, the Sanger method, with its modifications, eventually became the favored approach, overshadowing others until the advent of Next Generation Sequencing (NGS) around 2005.
The Birth of Genomics: Sanger Sequencing
Genomics is a multidisciplinary field of biological science that focuses on the study of the entire set of genes within an organism’s DNA, collectively known as its genome. It involves the comprehensive analysis and characterization of the structure, function, and organization of genes, as well as their interactions and regulation within the genome. The genome contains all the hereditary information needed for an organism’s growth, development, functioning, and reproduction. Genomics aims to decode and understand the genetic content of organisms, ranging from simple viruses and bacteria to complex plants, animals, and humans.

In 1977, Sanger and colleagues achieved a milestone by sequencing the first DNA-based genome, a <X174 bacteriophage, approximately 5375 base pairs long. The original Sanger sequencing method employed a two-phase DNA synthesis reaction. In the first phase, a DNA polymerase partially extended a primer on a single-stranded DNA template, generating DNA fragments of random lengths. In the second phase, these fragments were split into four parallel DNA synthesis reactions, each missing one of the four deoxyribonucleotide triphosphates (dNTPs). Consequently, the synthesis reactions stopped just before the missing base was incorporated. Electrophoresis on an acrylamide gel separated these fragments, and their sequence was read off a radioautograph, as they were synthesized with radiolabeled nucleotides (e.g., S-dATP).
The early Sanger method faced several challenges, including cumbersome two-phase procedures, limited sequence length determination, the need for known template sequences for primer design, the use of hazardous radioactive nucleotides, and lack of automation. Improvements followed rapidly, as Sanger and colleagues introduced dideoxynucleotides as chain terminators, simplifying the method to use single-phase reactions with fluorescent dyes. Each of the four dideoxynucleotides was labeled with a different fluorescent dye color, allowing for all four terminations to be analyzed on a single lane of the acrylamide gel. Moreover, the electrophoresis was coupled to a fluorescent detector connected to a computer, enabling automatic data collection. This innovation led to the commercialization of the first automated DNA sequencer in 1986 (i.e., Model 370A) by Applied Biosystems.
Overcoming Sequence Length Limitations: Shotgun Sequencing
While Sanger sequencing was a significant step forward, its read length was limited to around 500–800 bases, inadequate for analyzing most genes and complex DNA sequences. To address this, shotgun sequencing emerged as a powerful approach. In this method, longer DNA fragments are randomly sheared and cloned for sequencing. A computer program then assembles the sequences by identifying overlapping regions, creating a contiguous sequence or “contig.”
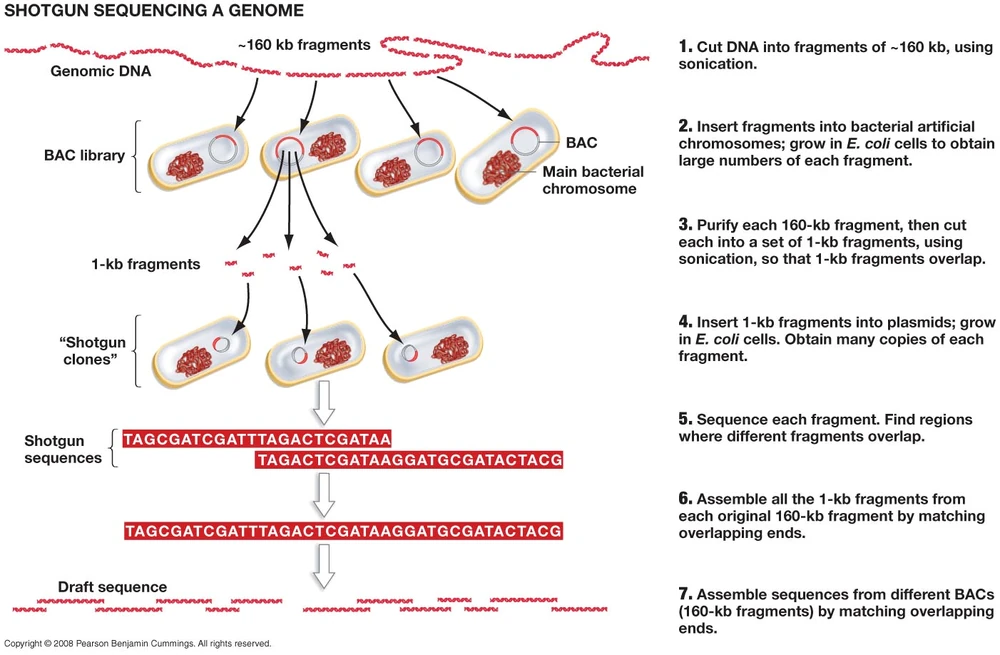
As the quantity of generated DNA fragments grew into the thousands or millions, aligning and assembling them became a formidable challenge. Early work in this area introduced alignment algorithms such as the Needleman–Wunsch and Smith–Waterman algorithms.
Needleman-Wunsch Algorithm: Global Sequence Alignment. The Needleman-Wunsch algorithm, developed by Saul Needleman and Christian Wunsch in 1970, is a dynamic programming algorithm used for global sequence alignment. Global alignment is employed to find the best alignment between two sequences, considering the entire length of both sequences and penalizing gaps and mismatches. It is commonly applied to compare DNA or protein sequences to identify evolutionary relationships and conserved regions. The algorithm constructs an alignment matrix by comparing each position in one sequence with every position in the other sequence. It assigns scores to matches, mismatches, and gap openings and extensions. The matrix is then filled in a systematic manner, starting from the top-left corner and moving towards the bottom-right corner. The optimal alignment is found by tracing back through the matrix, following the highest scoring path.
The Needleman-Wunsch algorithm guarantees the optimal global alignment by considering all possible alignments and choosing the one with the maximum score. However, this thoroughness makes it computationally expensive for large sequences, limiting its application to short sequences or small databases.
Smith-Waterman Algorithm: Local Sequence Alignment. The Smith-Waterman algorithm, developed by Temple F. Smith and Michael S. Waterman in 1981, is a dynamic programming algorithm used for local sequence alignment. Unlike global alignment, local alignment focuses on finding the best alignment within subsequences of the two sequences, allowing for gaps and mismatches outside these regions. This flexibility makes the Smith-Waterman algorithm ideal for detecting short similar regions within large sequences, such as identifying conserved domains or functional motifs in proteins. The algorithm employs a similar approach to the Needleman-Wunsch algorithm, creating an alignment matrix and filling it based on scores for matches, mismatches, gap openings, and extensions. However, the key distinction lies in the traceback process. Instead of tracing back from the bottom-right corner, the Smith-Waterman algorithm identifies the cell with the highest score in the matrix and traces back from there, following the highest scoring path until a zero-score cell is reached.
The Smith-Waterman algorithm excels at identifying local similarities, even in sequences with dissimilar overall composition. Its ability to pinpoint specific conserved regions makes it valuable in many bioinformatics applications, such as detecting homologous protein domains or identifying mutations within genes associated with diseases.
With the continued progress in bioinformatics and the advent of NGS, the shotgun sequencing approach revolutionized genomics, enabling scientists to decipher genomes with unprecedented speed and efficiency.
Genomics Onwards
The history of DNA sequencing is a tale of relentless pursuit, innovation, and technological breakthroughs. From the seminal discoveries of the DNA structure and genetic code by Watson, Crick, Wilkins, Holley, Khorana, and Nirenberg to the advent of Sanger sequencing and its eventual transformation into Next Generation Sequencing, each milestone has propelled genomics into new frontiers. As sequencing technologies continue to advance, decoding the blueprint of life has become more accessible, promising profound insights into the intricacies of existence and unlocking the secrets of living organisms.
Engr. Dex Marco Tiu Guibelondo, BS Pharm, RPh, BS CpE
Editor-in-Chief, PharmaFEATURES

Subscribe
to get our
LATEST NEWS
Related Posts

Bioinformatics & Multiomics
Data Deluge: Why Biomedical Informatics Must Reengineer Itself for the Era of Scientific Big Data
Biomedical big data is the scientific infrastructure that turns massive biological and clinical information streams into actionable medical knowledge.

Bioinformatics & Multiomics
Network Medicines: How AI is Teaching Small Molecules to Think in Pathways
AI-driven polypharmacology treats a small molecule not as a single-target bullet, but as a network-calibrated intervention designed for the real complexity of human disease.

Bioinformatics & Multiomics
Agentic Bioinformatics: How Autonomous AI Agents Compress Biomedical Discovery Cycles
Agentic bioinformatics treats biomedical discovery as a closed-loop system where specialized AI agents continuously translate intent into computation, computation into evidence, and evidence into the next experiment.

Bioinformatics & Multiomics
Proteomic Signatures: Molecular Discrimination of Hyperinflammatory States Through Serum Proteome Architecture
Serum proteomics exposes how sepsis and hemophagocytic syndromes diverge at the level of immune regulation and proteostasis, enabling precise molecular discrimination.
Read More Articles





Medicinal Chemistry & Pharmacology
April 14, 2026
Igor Nasonkin and Phythera Therapeutics: Moving Oncology Beyond Single Targets into Engineered Polypharmacologic Systems
Igor Nasonkin’s systems-driven approach at Phythera Therapeutics reframes oncology drug development from single-target inhibition to AI-enabled polypharmacologic network modulation using nature-derived molecular architectures.


Artificial Intelligence and Data Analytics
April 10, 2026
Inside Johnson & Johnson’s External Innovation Engine: Devin Swanson on Translating Integrated Discovery into Strategic Value
Devin Swanson’s leadership at Johnson & Johnson Innovative Medicines redefines external innovation as a tightly governed, AI-enabled translational system integrating multi-modal drug discovery, biomarker strategy, and capital-efficient execution.

Immunology & Oncology
April 9, 2026
From DMPK to Distributed Execution: Mehran F. Moghaddam’s Systems Strategy at OROX BioSciences, Inc.
A systems-level examination of how Mehran F. Moghaddam operationalizes DMPK, externalized R&D, and lipid-mediated therapeutics into a predictive, high-velocity biotech development architecture.