Measurement of Drug Concentrations
The centrality of identifying concentrations of drug in the body as key measure for personalized or group pharmacokinetics cannot be overstated. Biologic samples of these drug concentrations are measured from saliva, human breast milk, plasma, urine and even human fecal matter.
For the direct measurement of pharmaceuticals in biologic matrices, sensitive, accurate, and precise analytical approaches are available. These measurements are often validated in order to produce accurate data for pharmacokinetic and clinical monitoring. Generally speaking, chromatographic and mass spectrometric methods are used the most often to measure drug concentrations because chromatography separates the drug from other related substances that might interfere with assays and mass spectrometry enables the detection of molecules or molecules fragments based on their mass-to-charge ratio.
Sampling of Biologic Specimens
To learn more about the drug concentration in the body, only a small number of biologic specimens can be safely taken from the patient. Blood, spinal fluid, synovial fluid, tissue biopsy, and any biologic material that needs parenteral or surgical intervention on the patient are examples of invasive procedures. In contrast, noninvasive techniques involve taking samples of bodily fluids that don’t require parenteral or surgical assistance, such as urine, saliva, feces, expired air, or any other biological material.
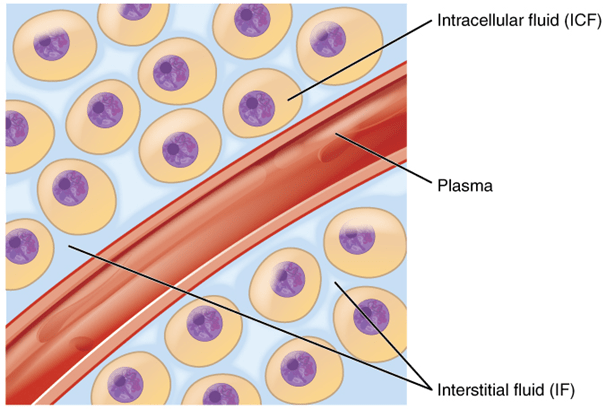
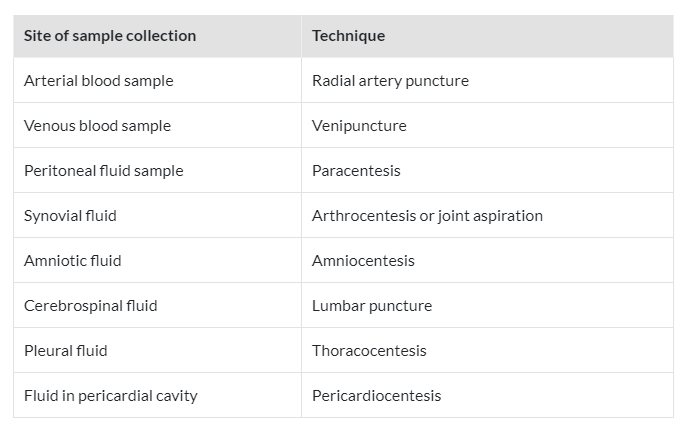
The amount of drug retained in or transported into that region of the tissue or fluid, the likelihood that drug dosing will have a certain pharmacologic or toxicologic effect, and the formation or transport of drug metabolites are all important details that can be learned by measuring the concentration of drug and metabolite in each of these biologic materials. Analytical techniques should be able to discriminate between parent drugs that are bound to proteins and those that are not, as well as between each metabolite, and they should be able to identify the pharmacologically active species. Particularly crucial for initial pharmacokinetic modeling of a drug are such discrepancies between metabolites in each tissue and fluid.
Drug Concentrations in Blood, Plasma, or Serum
The most straightforward method of determining how a medicine behaves in the body is to measure its concentrations (levels) in the blood, serum, or plasma. Red blood cells, white blood cells, platelets, and numerous other proteins, such as albumin and globulins, are among the biological components found in whole blood.
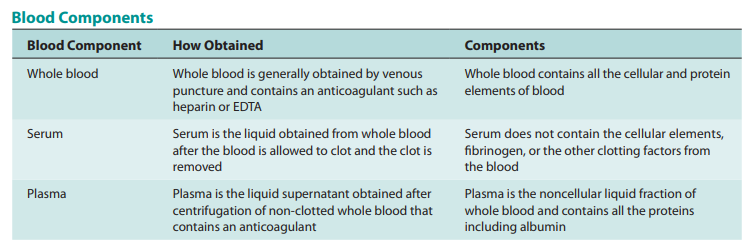
For measuring drugs, serum or plasma are most frequently utilized. Serum is obtained by allowing entire blood to coagulate, followed by centrifugation, then collection from the supernatant. The supernatant of centrifuged whole blood that has had an anticoagulant, such as heparin, added to it is used to make plasma. As a result, serum and plasma have different protein contents. All of the body’s tissues are perfused with plasma, including the blood’s cellular components.
If a drug in the plasma and tissues are in dynamic equilibrium, then changes in the plasma drug concentration will reflect changes in the tissue drug concentrations. Before measuring drug concentrations, plasma proteins are frequently removed since they are frequently attached to drugs in the plasma. This is the amount of unbound drug present. The total plasma drug concentration is the result of measuring the drug concentration from unfiltered plasma as an alternative. It’s crucial to know what kind of plasma concentration the data indicate when analyzing plasma concentrations.
Plasma Drug Concentration-Time Curve
After a drug product is provided, plasma samples are obtained at various time intervals, and the drug concentration in those samples is used to create the plasma drug concentration (level)-time curve. On Cartesian coordinate graph paper, the amount of drug present in each plasma sample is plotted against the precise moment at which the sample was taken. Plasma drug concentrations will increase to their maximum as the drug enters the general (systemic) circulation if it was administered intravenously. A medicine is often absorbed more quickly than it is excreted.
Medication distribution to all body tissues and elimination both occur simultaneously with drug absorption into the systemic circulation. A drug may be eliminated through excretion, biotransformation, or a combination of the two. Other methods of removal, such as those found in perspiration, exhaled air, and feces, may also be at play.
The graph below illustrates the link between the drug level-time curve and numerous pharmacologic characteristics of the substance.
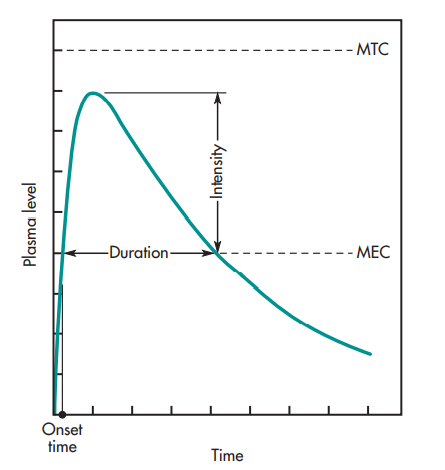
oral administration of a drug.
The terms “MEC” and “MTC” refer, respectively, to the minimal effective concentration and the minimum toxic concentration of a medicine. Knowing the drug concentration that barely has a pharmacologic effect is useful for some medications, such as those that affect the autonomic nervous system (i.e., MEC). The MEC is the lowest concentration of medication required at the receptors to provide the intended pharmacologic action, assuming that the drug concentration in the plasma is in equilibrium with the tissues. The MTC stands for the drug concentration required to barely cause a hazardous impact. The duration needed for the medication to get to the MEC is represented by the onset time.
Higher plasma drug concentrations induce a stronger pharmacologic response, up to a maximum, which is consistent with the notion that the intensity of the pharmacologic action is proportional to the number of drug receptors occupied. The time between the drug’s onset and its return to the MEC is known as the duration of action (DOA).
The concentrations between the MEC and the MTC are the therapeutic window. Generally speaking, medications having a broad therapeutic window are thought to be safer than those with a tight therapeutic window. The phrase “therapeutic index” is occasionally used. This phrase describes the proportion of toxic to therapeutic doses.
The pharmacokineticist can, however, also explain the plasma level-time curve using pharmacokinetic words like peak plasma level (Cmax), time for peak plasma level (Tmax), and area under the curve, or AUC (See figure below).
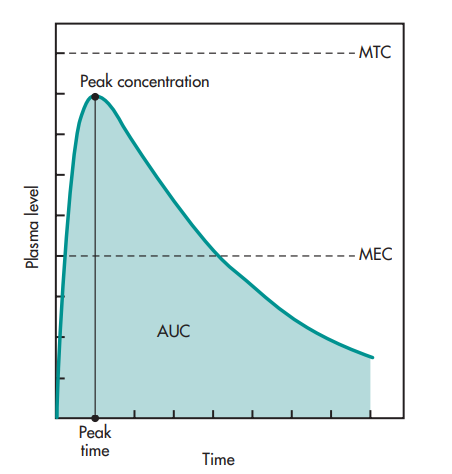
and concentration. The shaded portion represents the AUC
(area under the curve).
The greatest drug concentration in the plasma occurs at the peak plasma level, which serves as an approximate indicator of the typical rate of drug absorption. The dose, the absorption rate constant, and the drug’s elimination constant all affect the peak plasma level or maximum concentration. The amount of medication absorbed systemically is correlated with the AUC.
Drug Concentrations in Tissues
Occasionally, tissue biopsies are taken for diagnostic reasons, such as to confirm the presence of a malignancy. Measuring drug concentration is challenging since often just a small sample of tissue is extracted. Drug concentrations in tissue biopsies might not be an accurate representation of drug concentrations in other tissues or in all areas of the tissue from where the biopsy material was taken. For instance, the blood flow to the tumor cells might not be the same as the blood flow to other cells in this tissue if the tissue biopsy was performed to diagnose a tumor within the tissue.
In truth, blood flow need not be the same for all tissues for one section of the tissue to receive the same amount of blood as another part of the same tissue.
To determine whether the medicine entered the tissues and did so at the correct concentration, tissue biopsy material may be utilized to assess the drug concentration.
Drug Concentrations in Urine and Feces
The bioavailability of a medicine can be determined indirectly by measuring the substance in urine. The amount and pace of drug excretion in urine is a direct reflection of the amount and rate of drug absorption throughout the body.
Measuring a drug’s presence in feces may indicate that it was taken orally and not well absorbed, or it may indicate that it was absorbed systemically but was then evacuated by biliary secretion. When doing mass balance studies to account for the patient’s total dose, fecal medication excretion is frequently measured. Both urine and feces are collected, and the amount of drugs in each is determined for a mass balance study. Fecal collection is done to recover the dose form for some solid oral dosage forms that do not disintegrate in the gastrointestinal system but progressively leach out medication. The presence of any remaining medication is subsequently tested in the dose form that has not yet been dissolved.
Drug Concentrations in Saliva
For the purpose of therapeutic medication monitoring, the drug concentrations in saliva have been examined for numerous drugs. Since only free drugs diffuse into saliva, the concentration of free drugs in saliva is more likely to be close to the true value than the concentration of all drugs in plasma. For many medicines, the ratio of drug concentrations in the blood to those in the saliva is below 1. The medication’s pKa and salivary pH both have a significant impact on the ratio of drug concentrations in saliva and plasma. There is typically a greater association between plasma drug levels and weak acid and base medications with pKa values that differ significantly from pH 7.4 (plasma pH).
The indication of drug levels in the body is typically more stable when the saliva drug concentrations are measured after reaching equilibrium with the plasma drug concentration. It is best to use salivary medication concentrations as a secondary indicator or with caution as a treatment indicator.
Forensic Drug Measurements
The use of science in cases of murder, assault, and other crimes in court is known as forensic science. If a suspect or victim has taken an overdose of a prescription drug, has been poisoned, or has used illicit drugs like opiates (like heroin), cocaine, or marijuana, it may be helpful to measure the presence of drugs in the tissues obtained at autopsy or in other bodily fluids like saliva, urine, and blood. Drug examination of blood, urine, and saliva reveals short-term drug misuse by the presence of social drugs. Given their potential for fast elimination, it may be more challenging to demonstrate that the individual has been abusing narcotics.
Very sensitive test techniques, such gas chromatography and mass spectrometry, can analyze hair samples for drugs of abuse and provide information on previous drug exposure. Cocaine and 6-acetylmorphine, a heroin (diacetylmorphine) metabolite, were found in hair samples taken from patients who were known drug users, according to a study by Cone et al. (1993).
Significance of Measuring Drug Concentrations
The strength of the pharmacologic or poisonous action of the drug depends on the amount of the drug present at the receptor location, which is frequently located in the tissue cells. Because most tissue cells are extensively perfused with tissue fluids or plasma, measuring the plasma medication level is an efficient technique to monitor the success of therapy.
Individual differences in medication pharmacokinetics are quite common in clinical settings. Monitoring the drug concentration in the blood or plasma verifies that the dose delivered meets the necessary plasma level for therapeutic efficacy. In order to distinguish between patients who are taking too much of a drug and those who are extremely sensitive to it, monitoring of plasma levels is necessary for some drugs because receptor expression and/or sensitivity in people vary. Additionally, conditions such as disease, poor diet, the environment, concurrent pharmacological therapy, and other factors may have an impact on the patient’s physiologic functions. The link between plasma drug levels and pharmacologic response can be more precisely interpreted using pharmacokinetic models.
Plasma drug concentrations are largely worthless for dosage adjustment in the absence of pharmacokinetic data. A single blood sample from a patient, for instance, might be tested and shown to have 10 mg/mL. The highest safe dosage for this medicine is 15 mg/mL, according to the literature. When the blood sample was taken, what dosage of the medication was administered, and how it was administered must all be known in order to apply this information correctly. Pharmacokinetic equations and models may be utilized to precisely characterize the blood level-time curve and be used to change dose for that specific patient if the right data is provided.
For the purpose of customizing and maximizing therapeutic medication regimens, the monitoring of plasma drug concentrations enables dosage adjustments. Monitoring plasma medication concentrations may be able to give an indication of how the disease state is progressing when changes in physiologic functions occur, allowing the researcher to adjust the drug dosage as necessary. The most crucial factors from a clinical standpoint are good medical judgment and observation. Relying entirely on plasma medication concentrations for therapeutic decisions is not advised.
More often than not, it may be crucial to quantify the pharmacodynamic effects of the drug rather than just its plasma levels. For individuals using cardiotonic medications like digoxin, it’s crucial to check the electrophysiology of the heart, including an electrocardiogram (ECG). Prothrombin clotting time may show whether the correct dosage was obtained for an anticoagulant medication like dicumarol. The majority of diabetics who use insulin will check their own blood or urine glucose levels.
Plasma drug concentrations might not be a reliable indicator of pharmacodynamic response for medications that have an irreversible effect at the receptor site. Cancer chemotherapy drugs frequently prevent nucleic acid or protein biosynthesis in order to kill tumor cells. The pharmacodynamic response for these medications is not directly correlated with the plasma drug concentration. In this instance, the patient’s other pathophysiologic parameters and side effects are tracked to avoid harmful toxicity.
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Editor-in-Chief, PharmaFEATURES

Subscribe
to get our
LATEST NEWS
Related Posts

Medicinal Chemistry & Pharmacology
Igor Nasonkin and Phythera Therapeutics: Moving Oncology Beyond Single Targets into Engineered Polypharmacologic Systems
Igor Nasonkin’s systems-driven approach at Phythera Therapeutics reframes oncology drug development from single-target inhibition to AI-enabled polypharmacologic network modulation using nature-derived molecular architectures.

Medicinal Chemistry & Pharmacology
Drugging mRNA with Small Molecules: David Weitz on Platform Strategy at Syrna Therapeutics
David Weitz of Syrna Therapeutics explores how small molecule modulation of mRNA, enabled by AI-driven discovery and platform-centric execution, is redefining the boundaries of druggable biology.

Medicinal Chemistry & Pharmacology
Leaner Molecules: Structural Simplification Outperforming Brute-Force Potency Potency Chasing in Lead Optimization
Structural simplification is the science of turning chemically overbuilt leads into more efficient, drug-like molecules without surrendering their therapeutic logic.

Read More Articles






Artificial Intelligence and Data Analytics
April 10, 2026
Inside Johnson & Johnson’s External Innovation Engine: Devin Swanson on Translating Integrated Discovery into Strategic Value
Devin Swanson’s leadership at Johnson & Johnson Innovative Medicines redefines external innovation as a tightly governed, AI-enabled translational system integrating multi-modal drug discovery, biomarker strategy, and capital-efficient execution.

Immunology & Oncology
April 9, 2026
From DMPK to Distributed Execution: Mehran F. Moghaddam’s Systems Strategy at OROX BioSciences, Inc.
A systems-level examination of how Mehran F. Moghaddam operationalizes DMPK, externalized R&D, and lipid-mediated therapeutics into a predictive, high-velocity biotech development architecture.

{kind=link}
{kind=link}
{kind=link}