Next-generation sequencing (NGS) technologies have transformed genomics research by providing high-throughput sequencing capabilities with unparalleled speed and accuracy. The generated sequencing reads hold the key to unlocking valuable insights into genetic variations and disease mechanisms. However, the raw sequencing data from NGS platforms requires sophisticated computational analysis to extract meaningful information. This secondary analysis, which involves reads alignment and variant calling, is a critical step that bridges the gap between raw data and actionable genetic insights. In this article, we delve into the intricacies of secondary NGS analysis, exploring the process of read alignment, variant calling, and structural variant detection. By understanding the complexities and nuances of these analysis techniques, researchers can make informed decisions to harness the full potential of NGS data.
Reads Alignment: Mapping the Genetic Puzzle
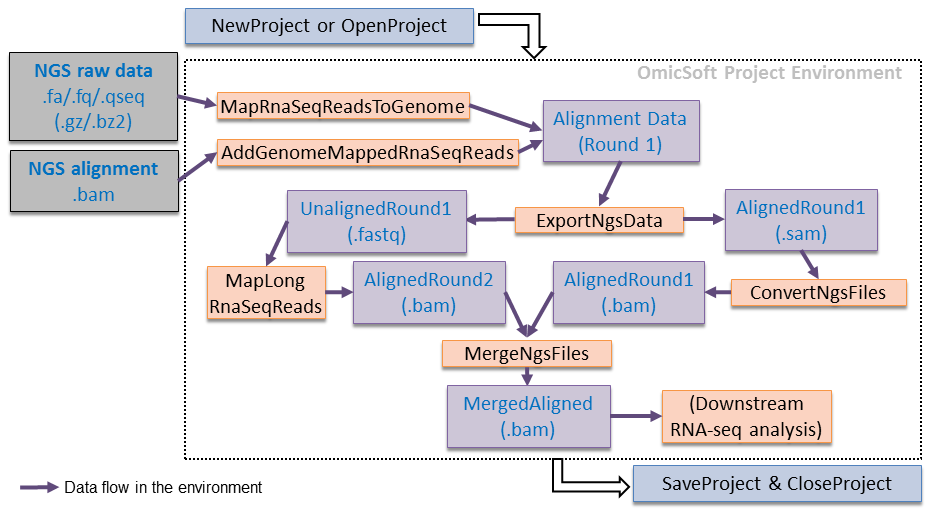
Reads alignment is the foundational step in secondary NGS analysis, where the sequenced fragments are aligned against a reference genome. This process is vital for accurately determining the genetic variants present in the sequenced samples. Researchers have two primary alternatives for reads alignment: read alignment against a reference genome or de novo assembly.
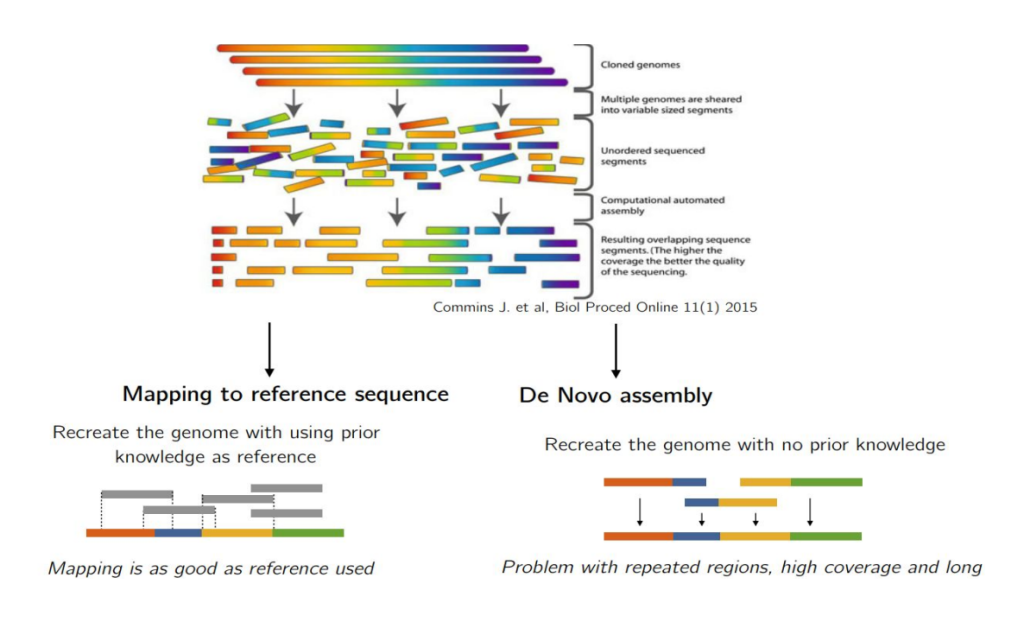
Read Alignment Against a Reference Genome
The process of read alignment against a reference sequence is the cornerstone of secondary NGS analysis, especially in clinical genetics. This approach is widely preferred due to its ability to efficiently identify genetic variants in the sequenced samples by comparing them to a known reference genome. By leveraging a reference genome, researchers can pinpoint the specific locations of variations within the sample, providing valuable insights into the genetic makeup of individuals.
To perform read alignment, sophisticated mapping algorithms are employed. These algorithms analyze the sequenced reads and attempt to find the most suitable position in the reference genome that closely matches each read. Since genetic variations are inherent in populations, the aligner allows for a certain number of mismatches or small insertions and deletions (INDELs) between the read and the reference sequence. This tolerance is necessary to accommodate the natural genetic diversity among individuals and accurately identify variants that may deviate slightly from the reference genome.
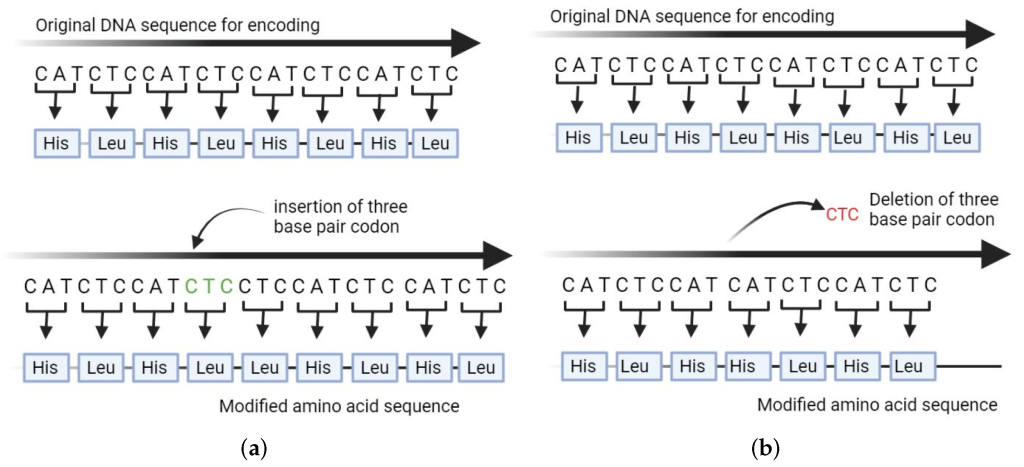
The aligner’s ability to tolerate variations is crucial, as genetic differences between individuals can manifest in various forms, such as single-nucleotide polymorphisms (SNPs) or small INDELs. By allowing for a controlled degree of variation, the aligner can still confidently identify these subtle genetic differences without compromising the accuracy of the analysis.
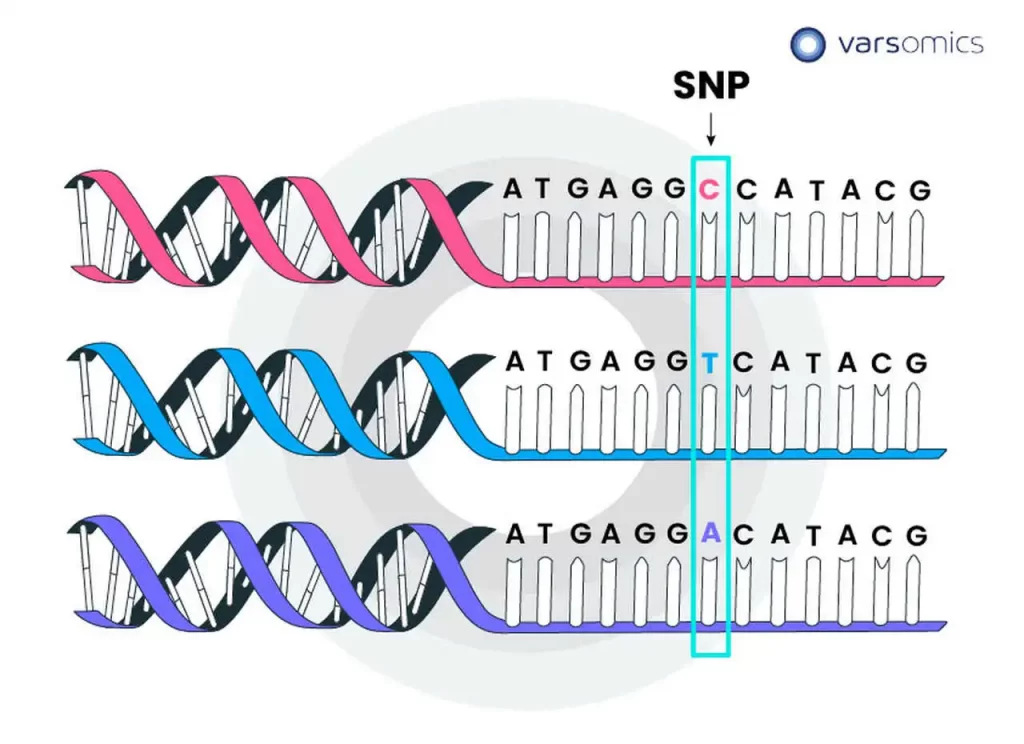
In clinical genetics, read alignment against a reference sequence is particularly advantageous for identifying disease-causing mutations and genetic variants associated with specific conditions. By comparing the sequenced reads to a well-established reference genome, researchers can pinpoint the genetic changes responsible for diseases or predict an individual’s susceptibility to certain conditions.
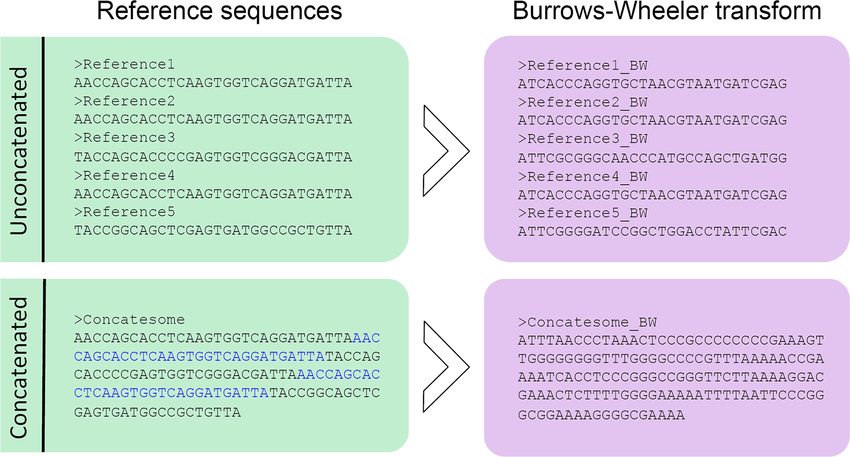
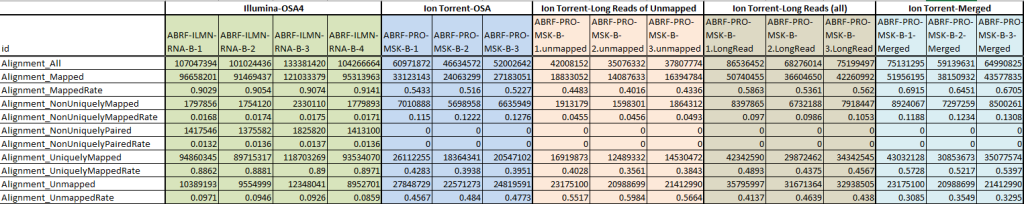
Short-read alignments are a critical aspect of NGS data analysis, and several powerful tools have been developed to efficiently align short DNA or RNA fragments to a reference genome. Among these tools, two popular options are Burrows-Wheeler Aligners (BWAs) and Bowtie (now Bowtie 2).
BWAs stand out for their ability to handle both short and long reads effectively. They leverage the Burrows-Wheeler transform algorithm, initially developed for data compression, to quickly align sequencing reads to the reference genome. The advantage of BWAs lies in their versatility, allowing researchers to process a wide range of read lengths with high efficiency. Whether dealing with short or long reads, BWAs offer reliable and accurate alignment results, making them a popular choice for various NGS applications.
On the other hand, Bowtie (now Bowtie 2) is renowned for its exceptional speed during alignment, making it particularly suitable for specific types of alignment tasks. However, the trade-off for this speed is the potential compromise on accuracy and sensitivity, especially when configured for maximum speed. As a result, Bowtie is more commonly used in RNA sequencing experiments, where speed is of utmost importance, and the focus is primarily on identifying gene expression levels rather than detecting genetic variants.
For data generated from the Ion Torrent platform, a specialized tool called the Torrent Mapping Alignment Program (TMAP) is recommended. TMAP is purposefully optimized to address the unique characteristics of Ion Torrent sequencing data. The alignment process in TMAP is executed in two stages to ensure the highest possible accuracy. In the initial mapping phase, TMAP uses algorithms like Smith-Waterman or Needleman-Wunsch to roughly align the reads against the reference genome. These algorithms are known for their ability to handle gapped alignments, which is crucial for accurately aligning reads with INDELs or other variations.
Following the initial mapping, TMAP proceeds with an alignment refinement step. This phase is designed specifically to compensate for specific systematic biases that are inherent in Ion Torrent sequencing. For example, Ion Torrent sequencing can introduce homopolymer alignment and phasing errors with low indel scores, and the alignment refinement in TMAP corrects for these potential errors. By accounting for these biases and fine-tuning the alignments, TMAP ensures that the resulting alignments are reliable and suitable for downstream analyses.
De Novo Assembly
In contrast to read alignment against a reference genome, de novo assembly involves reconstructing a genome from scratch without relying on an external reference. This approach is particularly useful when dealing with long-read sequencers such as those by Pacific Biosciences and Oxford Nanopore. De novo assembly can circumvent biases associated with a reference genome and improve the detection of structural variants (SVs) and complex rearrangements.
However, de novo assembly presents computational challenges, especially with short read lengths and highly repetitive regions in the genome. Assembling a genome from billions of DNA/RNA fragments, like a puzzle, requires sophisticated algorithms based on graphs theory. While de novo assembly holds promise, its current use is more confined to specific projects, such as correcting inaccuracies in the reference genome and enhancing SV identification. A separate article will be devoted entirely to De Novo Assembly.
The Art of Sequence Alignment and Its Challenges
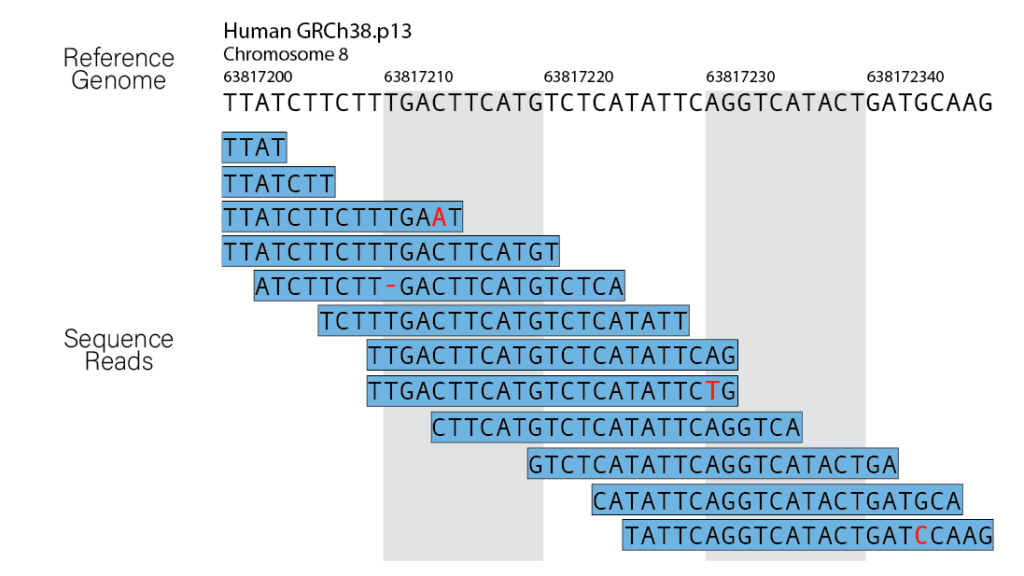
Sequence alignment, a classic bioinformatics problem, lies at the core of read alignment. The short read lengths generated by most NGS platforms demand the assembly of billions of DNA/RNA fragments, akin to piecing together a massive puzzle. However, challenges arise, especially when dealing with repetitive elements or regions where reads can belong to multiple copies of the genome.
Factors Contributing to Alignment Errors
Alignment errors in genomics refer to inaccuracies or mistakes that occur during the process of mapping sequenced reads to a reference genome. These errors can have significant implications for downstream analyses, particularly when identifying genetic variations, such as single nucleotide polymorphisms (SNPs), insertions, or deletions. Several factors contribute to alignment errors, and addressing them is crucial for obtaining reliable results in genomic studies.
Sequencing errors: Sequencing technology is not perfect, and errors can occur during the sequencing process, leading to discrepancies between the actual DNA sequence and the generated reads. These errors can introduce noise and misalignments during the mapping step.
Discrepancies with the reference genome: The reference genome used for mapping may not accurately represent the specific individual or population being studied. Genetic variations, such as insertions, deletions, or structural variants, can differ between individuals, and if the reference genome does not account for these differences, it can lead to misalignments.
Threshold determination: Differentiating true genetic variations from alignment artifacts can be challenging. Researchers need to establish appropriate thresholds for variant calling to minimize false positives and negatives. Setting these thresholds too strictly might lead to genuine variations being missed, while setting them too loosely may introduce false positives.
Data File Formats
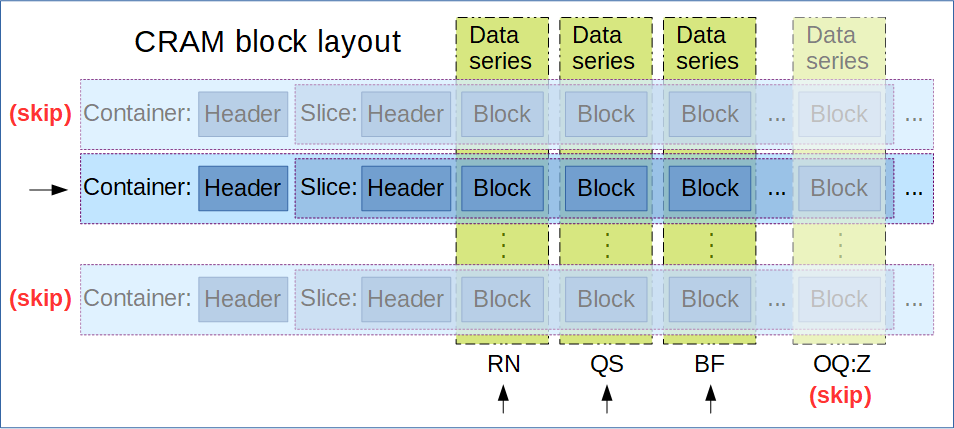
The widely accepted data input file format for assembly is FASTQ, while the output typically includes binary alignment/map (BAM) and sequence alignment/map (SAM) formats. These files contain critical information such as read sequence, base quality scores, alignment locations, differences relative to the reference sequence, and mapping quality scores (MAPQ).
Data Input File Format

FASTQ is a widely accepted and widely used data input file format for DNA sequencing data. It contains information about the sequences of DNA fragments obtained during the sequencing process, along with quality scores for each base call. FASTQ files typically consist of four lines per sequence:
Line 1: Starts with ‘@’ and contains a sequence identifier or read ID.
Line 2: The actual DNA sequence represented by a string of letters (A, T, C, G, or N for unknown bases).
Line 3: Starts with ‘+’ and is often used as an optional separator or repeat of the sequence identifier.
Line 4: Quality scores for each base call in Line 2. These scores represent the confidence or accuracy of the base call.
FASTQ files are essential for various bioinformatics applications, including genome assembly, variant calling, and quality control. The quality scores help researchers assess the reliability of each base call, aiding in error correction and filtering during the analysis.
Data Output File Formats
After obtaining sequencing reads, one of the essential tasks is to align these reads to a reference genome or assembly. The output of this alignment process is typically stored in either SAM or BAM format.
SAM Format: The Sequence Alignment/Map (SAM) format is a text-based file format that represents the aligned sequencing reads. It contains essential information, including the read ID, alignment location on the reference genome, CIGAR string (which represents the alignment operations and differences relative to the reference sequence), and mapping quality (MAPQ) scores.

BAM Format: The Binary Alignment/Map (BAM) format is a binary version of the SAM format. It is more compact and faster to process than the text-based SAM format. BAM files are generated by converting SAM files into a binary representation, which reduces file size and allows for efficient storage and retrieval of alignment data.
Both SAM and BAM files provide critical information for downstream analyses, such as variant calling, gene expression quantification, and structural variant detection. The alignment information in these files allows researchers to determine the location and characteristics of each read relative to the reference genome or assembly. Additionally, the mapping quality scores (MAPQ) in these files indicate the confidence of the alignment, helping researchers assess the reliability of the aligned data.
CRAM Format: The Compressed Reference-oriented Alignment Map (CRAM) format is a DNA sequence alignment file format developed by the European Bioinformatics Institute, offering higher compression compared to BAM and SAM formats. Unlike BAM and SAM, CRAM uses reference-based compression, which means it only stores base calls that differ from a specified reference sequence.
Ensuring Accuracy: Post-Alignment Processing
Before variant calling, post-alignment processing is crucial to increase variant call accuracy and improve downstream analyses. This step involves filtering out duplicate reads, realigning reads near INDELs, and recalibrating base quality scores.
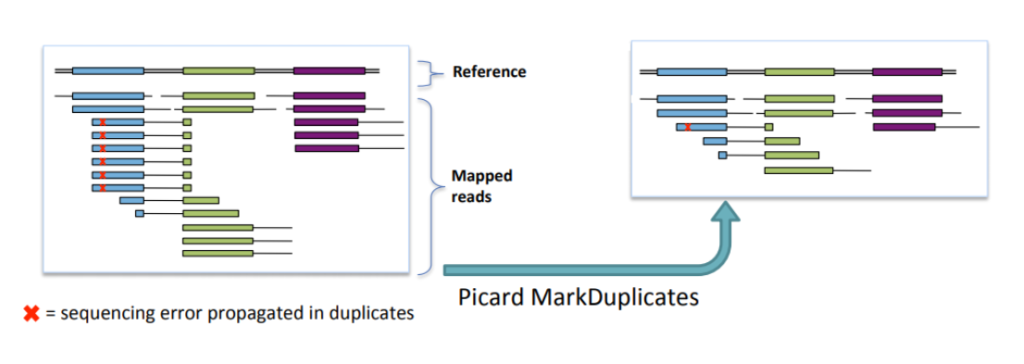
PCR duplicates and non-unique alignments must be removed to ensure that each fragment is treated independently during variant calling, preventing incorrect variant calls and inaccurate coverage and genotype assessments. Algorithms like MarkDuplicates in Picard tools are commonly used for this purpose.

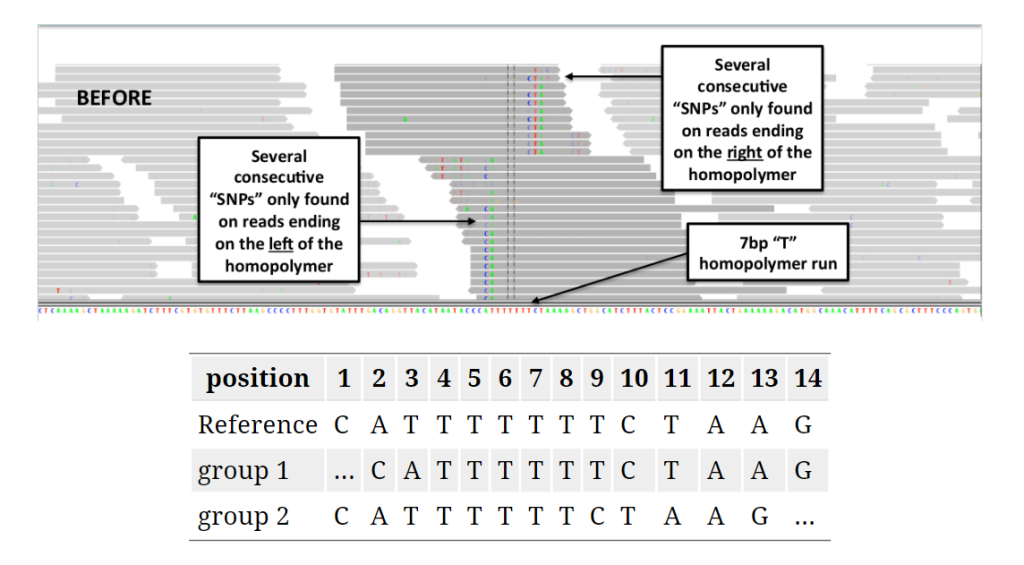
Reads spanning INDELs pose additional challenges, as the presence of an INDEL in a read can lead to alignment mismatches. Realigners like IndelRealigner from the GATK suite identify intervals requiring realignment due to INDELs and then perform a realignment to generate a consensus score supporting the presence of the INDEL.
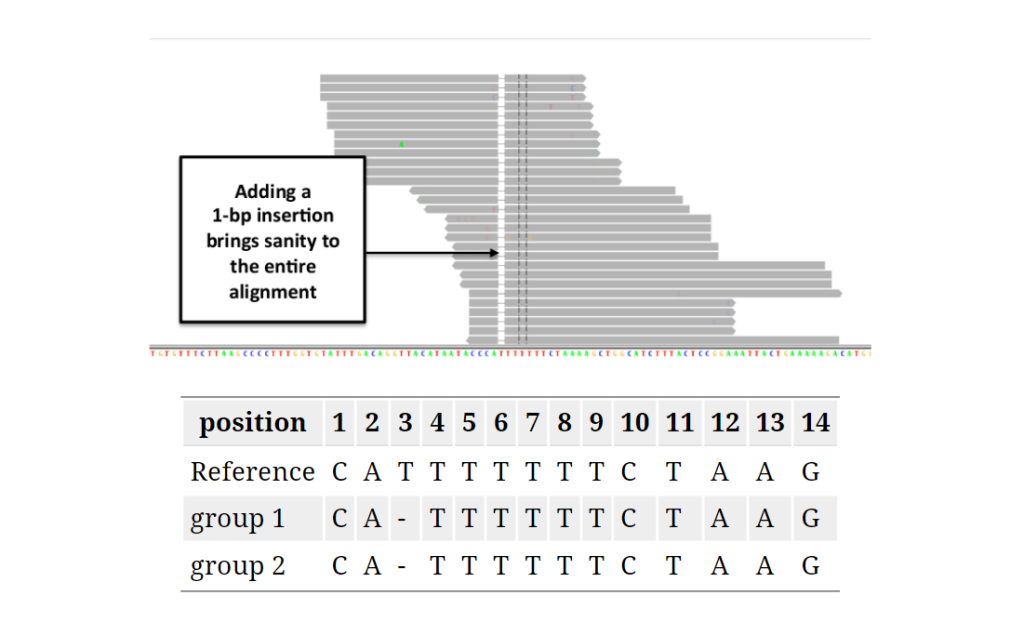

Base quality score recalibration is essential to refine the accuracy of base calls. Sequencing machines generate raw quality scores, but these may be influenced by various factors. By recalibrating base quality scores using tools like BaseRecalibrator from GATK, researchers can improve variant calling accuracy.
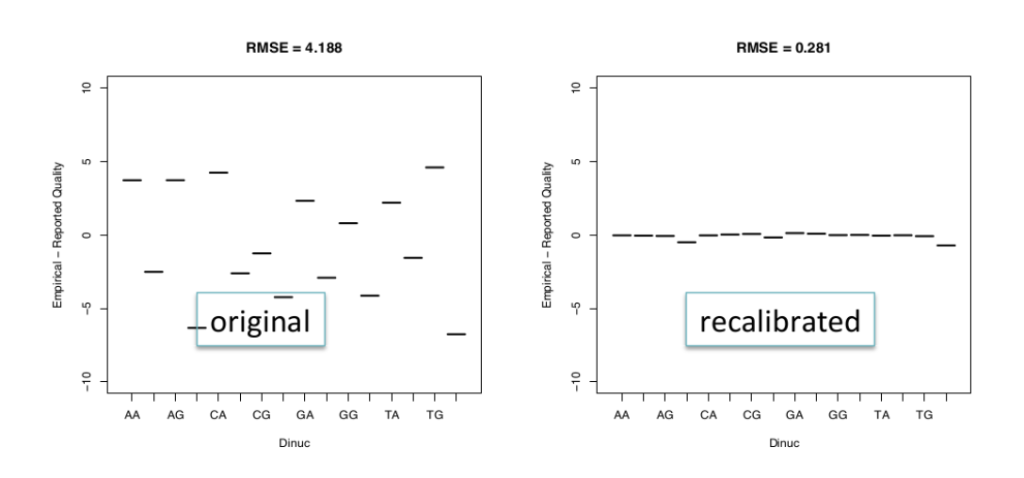


Post-Alignment Processing Methodologies
As mentioned, researchers employ post-alignment processing techniques to improve the accuracy of variant calling and reduce alignment artifacts. Some common methods include:
Base quality score recalibration: This process involves re-evaluating the quality scores assigned to each base in the sequencing data. It helps correct for biases in base calling and improves the accuracy of variant calling.
Local realignment: In regions with small insertions or deletions (indels), local realignment is performed to correct misalignments and produce more accurate alignments around these regions.
Duplicate read removal: During the sequencing process, some fragments of DNA may be duplicated, leading to duplicate reads. Removing these duplicates reduces the chance of misinterpretation and improves variant calling accuracy.
Variant quality score recalibration: Similar to base quality score recalibration, this step involves re-evaluating the variant quality scores, which helps in distinguishing true variants from false positives.
Filtering low-quality variants: Applying filters based on various metrics, such as variant depth, mapping quality, and strand bias, can help remove low-confidence variants from the final variant call set.
By carefully addressing these alignment errors and employing appropriate post-alignment processing steps, researchers can enhance the accuracy of variant calling, leading to more reliable and meaningful results in genomics studies.
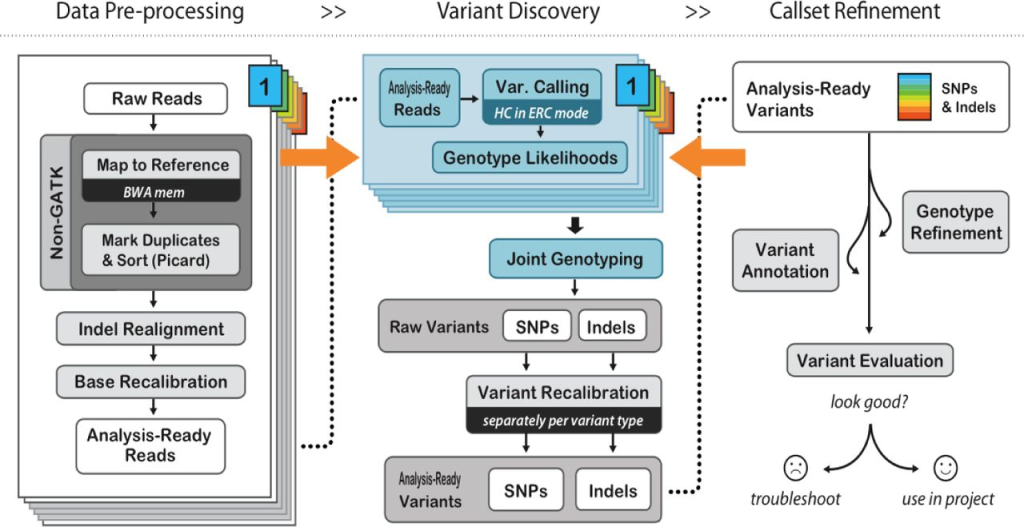
Variant Calling: Deciphering the Genomic Variations
Variant calling is the crux of secondary NGS analysis, where researchers identify genetic variants using post-processed BAM files. Several tools are available for variant calling, each utilizing different statistical methods to identify variant differences based on factors like base and mapping quality scores.
SAMtools, GATK (Genome Analysis Toolkit), Freebayes, and Torrent Variant Caller (TVC) are all software tools widely used in genomics for variant calling – the process of identifying genetic variations, such as single nucleotide polymorphisms (SNPs) and small insertions or deletions (indels), from DNA sequencing data. Each tool employs different algorithms and methodologies to achieve this task.
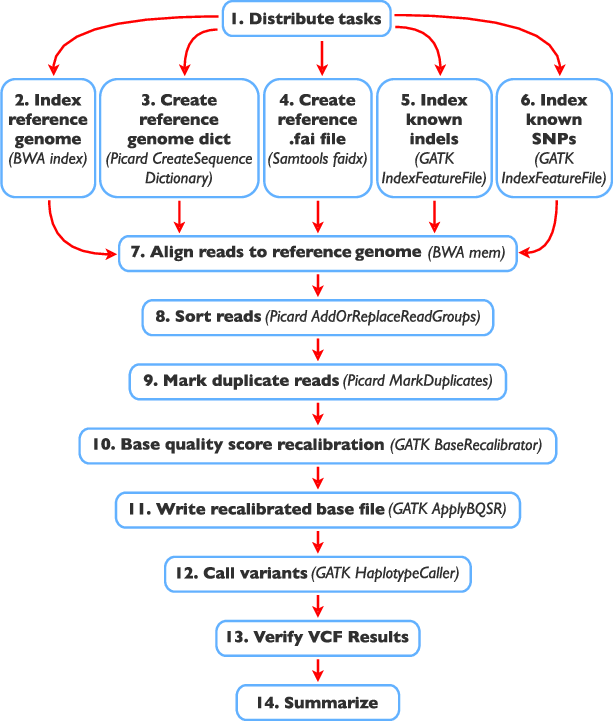
Tools Used to Implement Variant Calling
SAMtools: SAMtools is a popular and versatile software package used for processing and analyzing data in the SAM and BAM formats. It provides various functionalities for working with alignment data, including file format conversion, sorting, indexing, and manipulation of alignments. For variant calling, SAMtools implements algorithms based on the Bayesian statistical framework. One of its main functions for variant calling is “mpileup,” which evaluates the likelihood of different genotypes at each genomic position using sequencing data from multiple samples.
GATK (Genome Analysis Toolkit): GATK is a powerful and widely used software toolkit developed by the Broad Institute for variant discovery and genotyping. It is particularly popular in the research and clinical genomics community. GATK utilizes a combination of Bayesian and machine learning methods to call variants accurately and to provide reliable genotype likelihoods. It applies a step-by-step approach, which includes base quality score recalibration, local realignment around indels, and variant quality score recalibration, to improve the accuracy of variant calling. GATK is known for its robustness and sensitivity in detecting variants, especially in complex genomic regions.
Freebayes: Freebayes is another variant calling tool based on Bayesian statistical methods. It is designed to analyze diverse sequencing datasets, including pooled and population-scale samples. Freebayes uses Bayesian inference to call variants while considering variant and sequencing error probabilities. It leverages genotype likelihoods to estimate variant qualities and supports multiallelic calling. Freebayes is particularly efficient in detecting variants in situations where there is a high level of genetic diversity, such as in population-based studies.
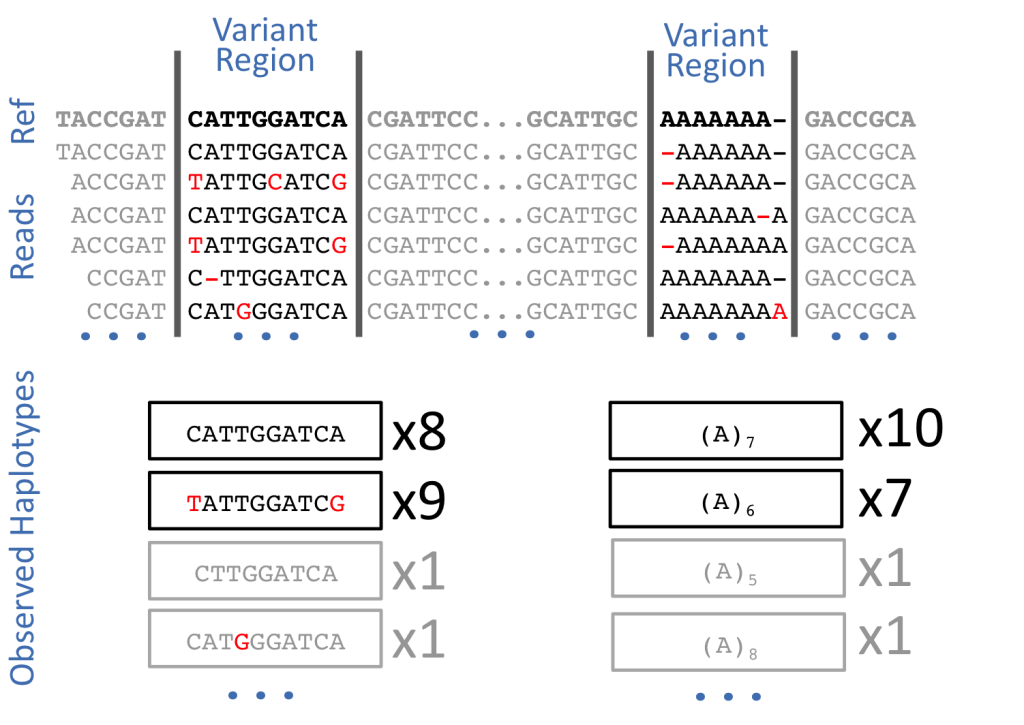
Torrent Variant Caller (TVC): TVC is a proprietary variant calling software specifically designed for data generated on the Ion Torrent sequencing platform. It is optimized to handle the unique characteristics of Ion Torrent sequencing data, such as the specific error profiles associated with this technology. TVC employs variant calling algorithms tailored to the Ion Torrent data format, allowing it to identify variants accurately and efficiently from the sequencing reads.
All of these tools take SAM/BAM files, which contain aligned sequencing reads, as input. After processing the data using their respective algorithms, they produce Variant Calling Format (VCF) files as output. VCF is a standard file format used to represent genetic variants and their associated information, such as chromosome location, variant type, quality scores, and genotypes. VCF files are commonly used for downstream analyses, population genetics studies, and clinical variant interpretation.

Unraveling Structural Variants: Challenges and Innovations
Definition and Classification of Structural Variants
Structural variants (SVs) are genomic alterations that involve large-scale changes in the DNA sequence, ranging from a few hundred base pairs to several kilobases. These variations can have a profound impact on health and disease, as they can disrupt genes, regulatory regions, or chromosomal architecture. SVs are known to play a significant role in various genetic disorders, including developmental disorders, intellectual disabilities, cancer, and many others.
There are several types of structural variants:
Large insertions/duplications: These are segments of DNA that are inserted into or duplicated within the genome. They can result in gene dosage changes, affecting the expression level of genes, and potentially leading to altered phenotypes.
Deletions (copy number variants, CNVs): Deletions involve the loss of DNA segments from the genome. CNVs refer to variations in DNA copy number, where a specific region is either duplicated or deleted in comparison to the reference genome.
Inversions: Inversions occur when a DNA segment is reversed in orientation within the genome. This can lead to changes in gene regulation or potential disruption of gene function.
The accurate identification of structural variants is crucial for understanding the genetic basis of diseases and for providing patients with appropriate diagnoses and treatments. Traditional short-read sequencing technologies, like Illumina sequencing, have limitations in detecting large SVs due to the relatively short read lengths.
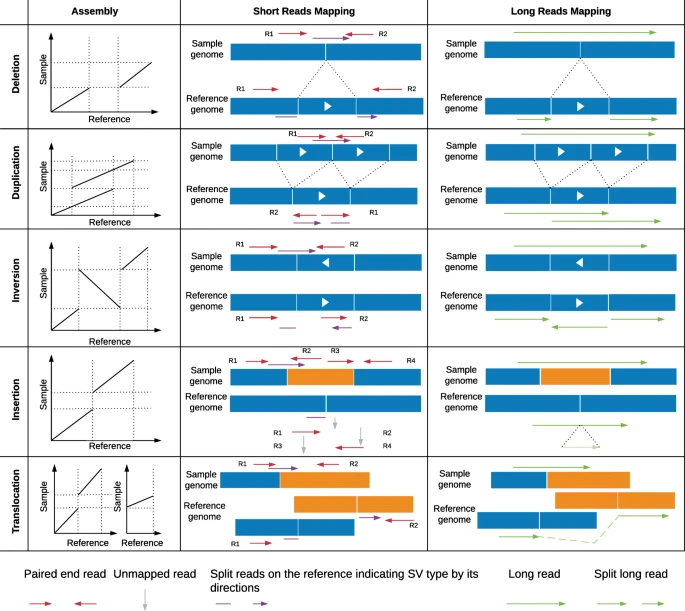
Advantages Afforded by Long-Read Sequencers
Third-generation sequencing technologies, such as Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), are referred to as long-read sequencers. These platforms generate much longer DNA sequence reads, spanning thousands of base pairs, compared to short-read sequencers. This makes them particularly well-suited for detecting large SVs, which are often challenging to identify using short-read data.
The advantages of long-read sequencers for identifying large SVs include:
Spanning repetitive regions: Long-read sequencers can traverse repetitive regions of the genome, which are prone to misalignments and assembly errors with short-read data. This ability improves the accuracy of SV detection in these regions.
Assembling complex SVs: Large SVs can be complex, involving multiple breakpoints and rearrangements. Long reads can span multiple breakpoints, aiding in the assembly of complex structural variations.
Detecting de novo SVs: Long-read sequencing is especially valuable for detecting de novo SVs in individuals without a reference genome or when studying species with limited genomic resources.
Approaches in Determining CNVs
Detecting copy number variants (CNVs) from next-generation sequencing (NGS) data is an important task in genomics research and clinical diagnostics. CNVs are genomic structural variations that involve gains or losses of DNA segments, and they can have significant implications for various diseases and conditions. Several strategies have been developed to identify CNVs from NGS data, each with its strengths and limitations. The choice of the detection method depends on the specific objectives of the study and the characteristics of the available data.
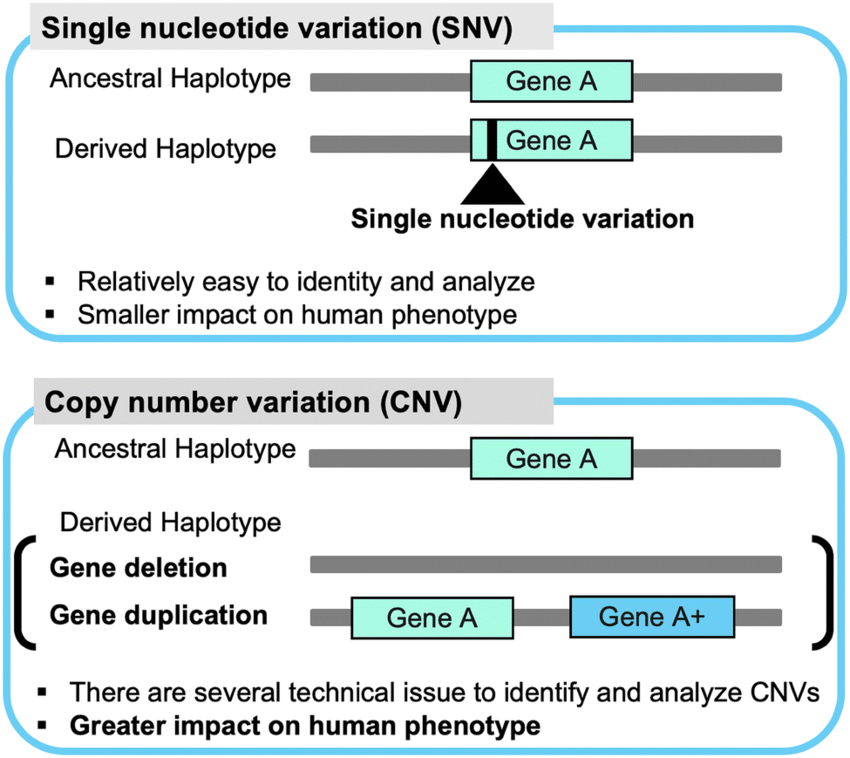
Paired-end mapping: Paired-end mapping is a commonly used approach for CNV detection. In this method, short reads are sequenced from both ends of DNA fragments, and the distances between the paired reads are used to estimate the fragment size. Deviations in the expected fragment size can indicate the presence of CNVs. This method is particularly effective for detecting large CNVs and is computationally efficient. However, it may struggle to accurately identify small or nested CNVs and regions with repetitive elements.
Split read: The split read method detects CNVs by identifying reads that partially align to different genomic locations, suggesting that a structural variation has disrupted the continuous alignment of the read. This approach is advantageous for detecting precise breakpoints of CNVs, including small insertions and deletions. However, it requires longer reads and can be computationally intensive.
Read depth: Read depth analysis estimates the number of reads that align to a specific genomic region, which can indicate copy number changes. Changes in read depth relative to the expected value for a diploid genome suggest the presence of CNVs. This method is effective for detecting large CNVs and can be applied to both short-read and long-read sequencing data. However, it may struggle with accurately identifying CNV breakpoints or distinguishing between different types of structural variations.
De novo genome assembly: De novo genome assembly involves reconstructing a complete genome sequence from short reads without relying on a reference genome. This approach can be useful for detecting CNVs in regions with no reference or for studying non-model organisms. However, de novo assembly is computationally demanding and requires high sequencing coverage for accurate results.
Combinatorial approaches: Many CNV detection tools use a combination of multiple strategies to increase accuracy and sensitivity. These methods integrate information from paired-end mapping, split read analysis, and read depth to identify CNVs. Combining multiple approaches can mitigate individual method limitations and improve overall CNV detection performance.
CNV detection from NGS data is a complex task, and different strategies exist to address various challenges. Researchers and clinicians need to consider the specific goals of their study, the types of CNVs they aim to detect, and the available data characteristics when choosing an appropriate method or employing a combination of approaches. Additionally, advances in sequencing technologies and bioinformatics tools continue to refine CNV detection methods, enhancing their accuracy and applicability in genomics research and clinical practice.
Ready for the Final Interpretation
Secondary NGS analysis is a multifaceted process that unlocks the potential of raw sequencing data by aligning reads against a reference genome, detecting genetic variants, and revealing structural variations. Understanding the intricacies of read alignment, variant calling, and SV detection is critical for researchers to make informed decisions and extract meaningful genetic insights from NGS data. As NGS technologies continue to evolve and computational tools advance, secondary analysis will remain at the forefront of cutting-edge genomics research, driving advancements in precision medicine and disease understanding.
Engr. Dex Marco Tiu Guibelondo, BS Pharm, RPh, BS CpE
Editor-in-Chief, PharmaFEATURES

Subscribe
to get our
LATEST NEWS




Related Posts

Bioinformatics & Multiomics
Mapping the Invisible Arrows: Unraveling Disease Causality Through Network Biology
What began as a methodological proposition—constructing causality through three structured networks—has evolved into a vision for the future of systems medicine.

Bioinformatics & Multiomics
Open-Source Bioinformatics: High-Resolution Analysis of Combinatorial Selection Dynamics
Combinatorial selection technologies are pivotal in molecular biology, facilitating biomolecule discovery through iterative enrichment and depletion.









