In the intricate world of pharmacodynamics, the fundamental principles governing drug actions are a captivating subject of study. To understand the profound influence that drugs exert on our bodies, one must delve into the multifaceted realm of drug-receptor interactions. This article embarks on an expedition through the captivating intricacies of pharmacodynamic principles, exploring various facets of these interactions, ranging from agonists to inverse agonists, and unraveling the mysteries behind the duration of drug action. We will also touch upon the significance of receptors and inert binding sites, shedding light on the essential elements that govern the efficacy and selectivity of these interactions.
Types of Drug-Receptor Interactions
At the heart of pharmacodynamics lies the concept that most drugs initiate their effects by binding to specific receptors. This initial binding serves as the inception of a complex cascade of events leading to the desired outcome. There are several distinct types of drug-receptor interactions, each with its unique characteristics.

Agonists
These remarkable molecules form a crucial category of drugs that bind to receptors and activate them, thereby instigating the desired effect. In many cases, the molecular structure of receptors undergoes a transformative change upon activation. Some receptors incorporate effector machinery within themselves, facilitating direct effects upon drug binding, such as the opening of ion channels or activation of enzyme activity. Others are interconnected with effector molecules through coupling molecules, which relay the signal downstream.
Pharmacologic Antagonists
In contrast to agonists, pharmacologic antagonists bind to receptors but block their activation, effectively competing with and preventing other molecules from binding. These antagonists, such as atropine for acetylcholine receptors, stabilize the receptor in an inactive state, diminishing the effects of agonist molecules. Nonetheless, their effects can be overcome by increasing the dosage of agonists. Some antagonists, when binding to the receptor site, form tight and irreversible bonds, making them impervious to displacement even with higher agonist concentrations. There are also allosteric interactions, where drugs bind to the same receptor but do not hinder agonist binding, instead enhancing or inhibiting the action of the agonist molecule.
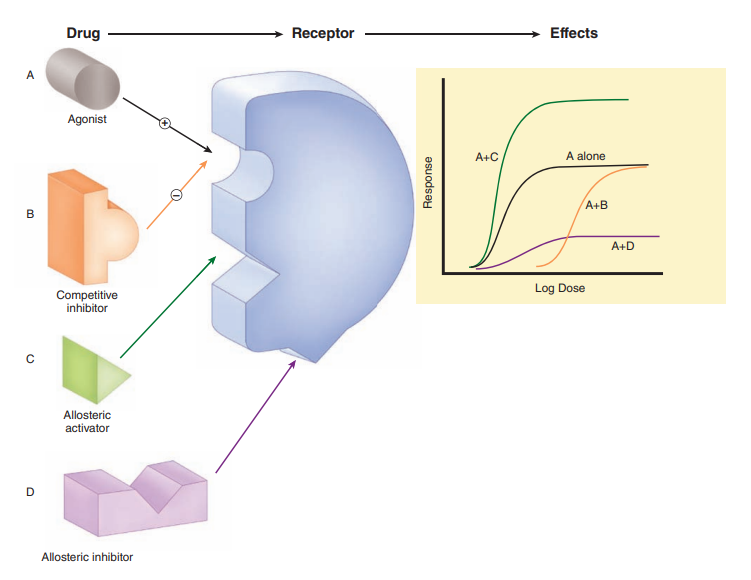
Agonists That Inhibit Their Binding Molecules
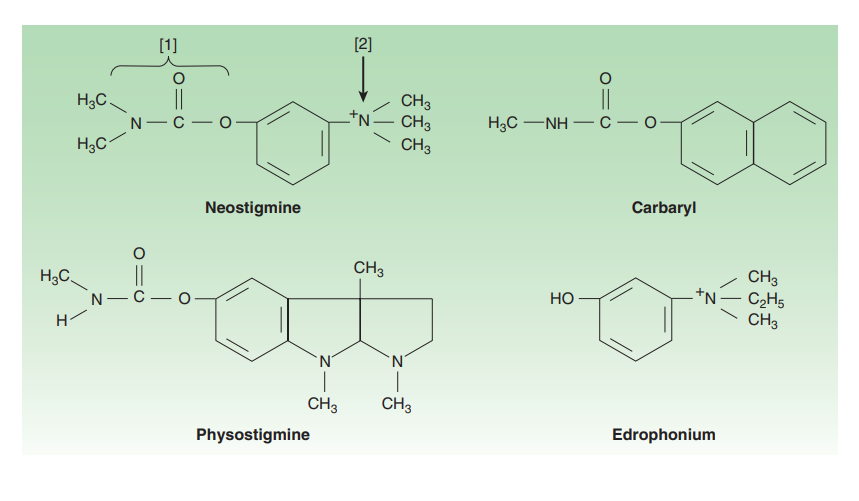
Certain drugs imitate agonists by inhibiting the molecules responsible for terminating the action of endogenous agonists. An exemplary case is acetylcholinesterase inhibitors, which impede the degradation of endogenous acetylcholine. While these inhibitors may not bind directly to cholinoceptors, their actions closely resemble those of cholinoceptor agonists. This mimicry amplifies the effects of naturally released agonist ligands, often leading to more selective and less toxic outcomes than exogenous agonists.
Agonists, Partial Agonists, and Inverse Agonists
A useful model emerges in the intricate dance of drug-receptor interaction, illustrating that receptors can exist in both an inactive (Ri) and an activated (Ra) state. Even without agonists, a fraction of receptors exist in the Ra form, generating constitutive activity. Agonists exhibit a higher affinity for the Ra configuration, stabilizing it and driving a substantial portion of receptors into the Ra-D (drug-activated receptor state) fraction, producing a significant effect. These full agonists can saturate receptor pools, activating them to their maximum capacity.
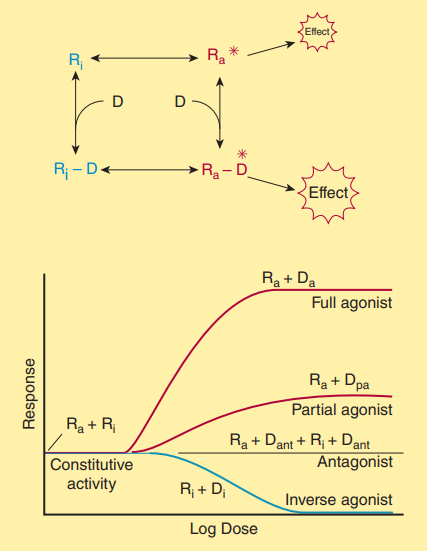
On the other hand, partial agonists, although binding to the same receptors and activating them similarly, evoke a less robust response, regardless of concentration. These partial agonists do not stabilize the Ra configuration as effectively as full agonists, resulting in a substantial fraction of receptors remaining in the Ri-D pool. This characteristic defines partial agonists as having low intrinsic efficacy. Importantly, they can also hinder access by full agonists, effectively acting as agonists in the absence of full agonists or antagonists when coexisting with full agonists.
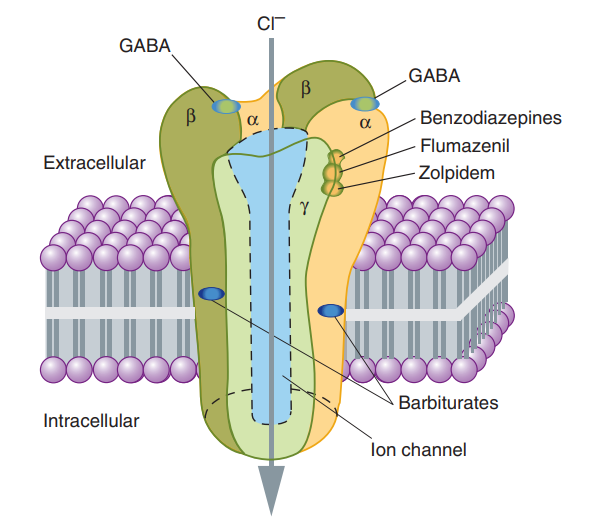
In instances where a drug exhibits a notably stronger affinity for the Ri state rather than the Ra state, leading to the stabilization of a substantial portion in the Ri-D pool, the resulting effect is a reduction in constitutive activity. This outcome stands in stark contrast to the effects induced by conventional agonists at the same receptor site. Drugs with such characteristics are referred to as inverse agonists. One of the most well-documented illustrations of this phenomenon is observed in the γ-aminobutyric acid (GABAA) receptor-effector system, which serves as a chloride channel within the nervous system.
The GABAA receptor responds to the endogenous transmitter GABA by promoting the inhibition of postsynaptic cells. Traditional exogenous agonists like benzodiazepines also enhance the functioning of the receptor-effector system, eliciting GABA-like inhibition that results in sedation, a therapeutic effect. This sedative effect can be reversed by conventional neutral antagonists such as flumazenil. However, when inverse agonists interact with this receptor system, they induce anxiety and agitation, which represents the opposite of sedation, elucidating the intriguing dynamics of drug-receptor interactions in this context.
Duration of Drug Action
Terminating drug actions involves several intricate processes. In some instances, effects persist only as long as the drug occupies the receptor, with dissociation of the drug from the receptor naturally concluding the effect. However, in many cases, actions continue after dissociation because coupling molecules remain activated. Covalently bound drugs may sustain effects until the drug-receptor complex disintegrates and new receptors or enzymes are synthesized. Many receptor-effector systems employ desensitization mechanisms to prevent excessive activation when agonists are continuously present.
Receptors and Inert Binding Sites
For a molecule to serve as a receptor, it must exhibit selectivity in ligand binding and change its function upon binding, thereby altering the function of the biologic system. The selectivity feature is vital to avoid constant activation by various ligands. Some endogenous molecules, despite their ability to bind drugs, do not function as regulatory molecules. Binding to nonregulatory molecules like plasma albumin, termed inert binding sites, may not alter biologic system function but significantly impacts drug distribution and the amount of free drug in circulation, thereby influencing pharmacokinetics.
In conclusion, the intricate landscape of drug-receptor interactions unfolds as a captivating tapestry of pharmacodynamic principles. From agonists to inverse agonists, these interactions orchestrate the delicate balance of physiological responses within the human body, offering insights into the mysteries of drug action. Understanding these principles is crucial for the development of effective and targeted therapeutic interventions, ultimately benefiting the field of medicine and the well-being of patients worldwide.
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Subscribe
to get our
LATEST NEWS
Related Posts

Medicinal Chemistry & Pharmacology
Spatial Collapse: Pharmacologic Degradation of PDEδ to Disrupt Oncogenic KRAS Membrane Localization
PDEδ degradation disrupts KRAS membrane localization to collapse oncogenic signaling through spatial pharmacology rather than direct enzymatic inhibition.

Medicinal Chemistry & Pharmacology
Neumedics’ Integrated Innovation Model: Dr. Mark Nelson on Translating Drug Discovery into API Synthesis
Dr. Mark Nelson of Neumedics outlines how integrating medicinal chemistry with scalable API synthesis from the earliest design stages defines the next evolution of pharmaceutical development.

Medicinal Chemistry & Pharmacology
Exelixis Clinical Bioanalysis Leadership, Translational DMPK Craft, and the Kirkovsky Playbook
Senior Director Dr. Leo Kirkovsky brings a rare cross-modality perspective—spanning physical organic chemistry, clinical assay leadership, and ADC bioanalysis—to show how ADME mastery becomes the decision engine that turns complex drug systems into scalable oncology development programs.
Read More Articles




Clinical Operations
February 3, 2026
Zentalis Pharmaceuticals’ Clinical Strategy Architecture: Dr. Stalder on Data Foresight and Oncology Execution
Dr. Joseph Stalder of Zentalis Pharmaceuticals examines how predictive data integration and disciplined program governance are redefining the future of late-stage oncology development.

Regulatory Affairs
January 30, 2026
Policy Ignition: How Institutional Experiments Become Durable Global Evidence for Pharmaceutical Access
Global pharmaceutical access improves when IP, payment, and real-world evidence systems are engineered as interoperable feedback loops rather than isolated reforms.

Artificial Intelligence and Data Analytics
January 29, 2026
Sepsis Shadow: Machine-Learning Risk Mapping for Stroke Patients with Bloodstream Infection
Regularized models like LASSO can identify an interpretable risk signature for stroke patients with bloodstream infection, enabling targeted, physiology-aligned clinical management.

Infectious Diseases & Vaccinology
January 28, 2026
Enduring Blockade: Five-Year Functional Antibody Persistence Against Emerging GII.4 and GII.17 Noroviruses
Natural infection with dominant GII noroviruses elicits long-lived functional antibodies, redefining immune durability in viral gastroenteritis.

Immunology & Oncology
January 27, 2026
Signal Switch: Stimuli-Responsive Nanoplatforms That Turn STING On Only Where Tumors Make Sense
Stimuli-responsive STING nanomedicine is an effort to make innate immune activation behave like a gated, tumor-local event rather than a body-wide inflammatory signal.