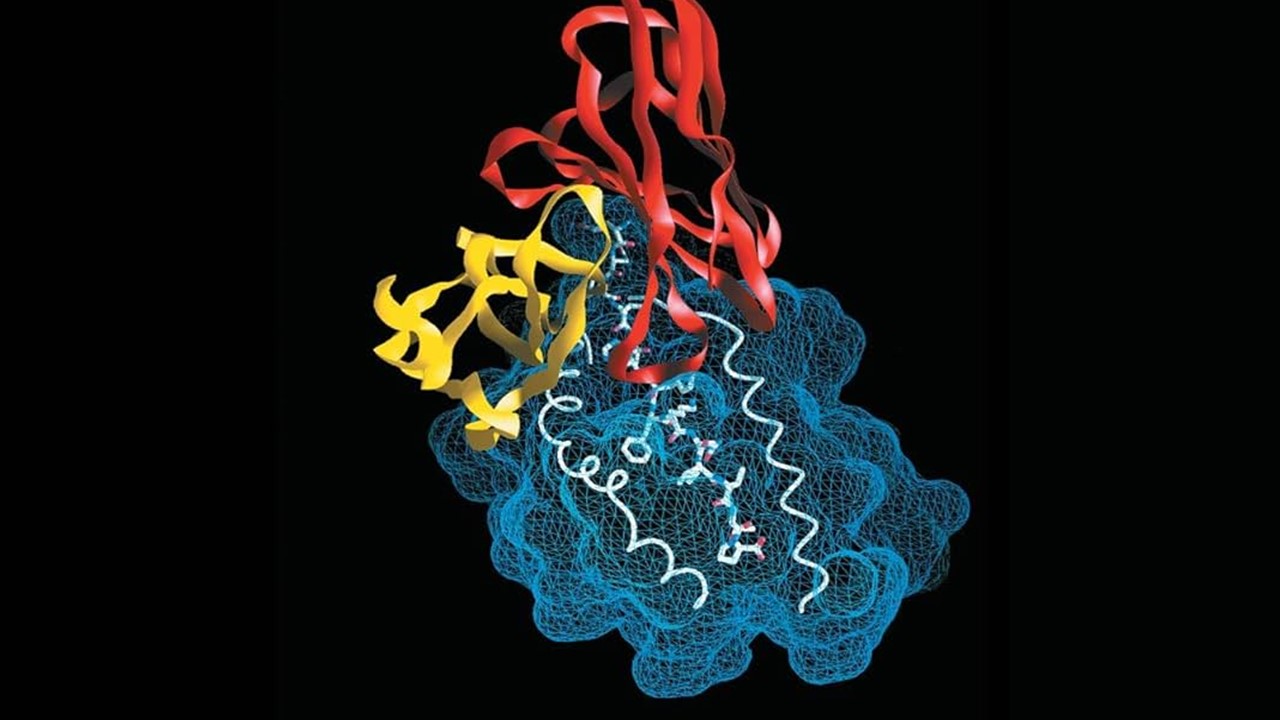
The BEDAM Method: A Computational Microscope for Molecular Interactions
At the heart of modern drug discovery lies the challenge of predicting how tightly a small molecule will bind to its target protein. The Binding Energy Distribution Analysis Method (BEDAM) emerges as a sophisticated computational tool designed to unravel this puzzle. By integrating Hamiltonian Replica Exchange Molecular Dynamics (HREM) with implicit solvent models like AGBNP2, BEDAM maps the thermodynamic landscape of ligand-protein interactions. This approach simulates a spectrum of states between fully coupled (bound) and decoupled (unbound) systems, allowing researchers to quantify binding free energies without relying on static crystal structures.
The FKBP12 system, a well-characterized immunophilin enzyme, served as the testing ground. Known for its shallow hydrophobic binding pocket, FKBP12 interacts with ligands ranging from modest fragments to the bulky immunosuppressant FK506. BEDAM’s ability to sample conformational space without rigid restraints revealed binding modes consistent with crystallographic data, validating its predictive power. The method’s reliance on multi-state free energy estimators, such as MBAR, ensures robust statistical analysis, merging data across λ-states to capture the full energetic spectrum of interactions.
A critical innovation in BEDAM is its “soft-core” binding energy function, which mitigates numerical instabilities caused by atomic clashes during alchemical transitions. By capping extreme repulsive forces, this modification enables smoother exploration of intermediate states, particularly crucial for large ligands like FK506. The result is a more accurate depiction of the binding process, where entropic penalties and enthalpic gains are balanced across a dynamic conformational ensemble.
Convergence Conundrums: When Algorithms Meet Biological Complexity
Convergence in free energy calculations is not merely a technical hurdle—it reflects the physical reality of molecular interactions. For smaller FKBP12 ligands, BEDAM achieved equilibrium between bound and unbound states, with replicas freely transitioning across λ-values. However, FK506—a macrocyclic behemoth—exposed the method’s limitations. Its rigid structure and extensive hydrophobic contacts created a stark energetic divide between states, slowing conformational transitions to a crawl.
This phenomenon mirrors protein folding’s order-disorder transitions. At critical λ-values, where bound and unbound populations are balanced, the system undergoes pseudo-phase transitions. The steepness of these transitions correlates with the ligand’s binding energy disparity: FK506’s large energy gap necessitated stricter restraints to confine sampling near the crystallographic pose. Without such constraints, replicas became trapped in disjointed energetic basins, stalling convergence.
The lesson is clear. While λ-hopping accelerates sampling of positional and rotational degrees of freedom, internal rearrangements—like sidechain rotations or loop movements—require bespoke strategies. For FK506, enforcing orientational restraints mimicked prior structural knowledge, enabling convergence. This underscores a trade-off: unrestrained sampling captures novel binding modes but risks computational intractability for complex systems.
Hydrophobic Forces: The Hidden Architects of Binding Affinity
Hydrophobic interactions often dominate ligand binding, yet modeling them remains fraught with challenges. The AGBNP2 implicit solvent model, while efficient, underestimated hydrophobic contributions in FKBP12 complexes. Its analytical surface area calculation failed to fully capture burial of loosely packed interfaces, misrepresenting solvent-exposed voids as hydrated regions. This led to systematic underestimation of binding affinities, particularly for larger ligands with extensive nonpolar surfaces.
A rescoring strategy salvaged accuracy. By replacing AGBNP2’s cavity term with a numerical solvent-accessible surface area (SASA) model, the hydrophobic driving force was recalibrated. The optimal surface tension coefficient aligned with independent studies on alkane hydration, affirming the physical basis of this correction. Post-hoc adjustments transformed BEDAM’s predictions, yielding binding free energies in striking agreement with experiments.
This work highlights a broader truth: implicit solvent models must balance computational speed with geometric precision. Current models excel at capturing electrostatic effects but falter in crowded hydrophobic interfaces. Future iterations could integrate machine learning to refine nonpolar terms, bridging the gap between rapid simulations and experimental fidelity.
Thermodynamic Tug-of-War: Energy vs. Entropy in Ligand Binding
Binding free energies are not mere sums of molecular interactions—they embody a contest between energetic gains and entropic losses. For FKBP12 ligands, the average binding energy (⟨u⟩₁) grew more favorable with ligand size, driven by additional hydrophobic contacts. Yet, this gain was partially offset by rising reorganization free energies (ΔG°reorg), reflecting the entropic cost of immobilizing flexible ligands and reshaping their conformations.
Smaller ligands, like compound 2, incurred modest reorganization penalties (~20 kcal/mol), allowing net favorable binding. In contrast, FK506’s rigid framework imposed a steep ~28 kcal/mol penalty, underscoring the paradox of molecular recognition: stronger interactions often demand greater conformational sacrifices. This duality complicates drug design, as optimizing affinity may inadvertently rigidify ligands, reducing bioavailability.
The data also revealed nonlinear trends. Ligands 6 and 8, despite similar sizes, exhibited divergent ΔG°reorg values, hinting at subtle structural influences on flexibility. Such nuances emphasize the need for dynamic sampling—static snapshots cannot capture the entropic tax imposed by molecular adaptation.
Binding and Unbinding: The Molecular Waltz of Ligand-Protein Dynamics
Ligand binding is seldom a lock-and-key mechanism. For FKBP12, BEDAM captured a stepwise dance: initial weak contacts preceded full engagement. Smaller ligands often adopted metastable intermediates, where one hydrogen bond formed while the ligand’s ester group dangled unanchored. Subsequent sidechain rotations snapped the second bond into place, akin to a latch securing a door.
These transitions peaked at λ ≈ 0.5, where bound and unbound states coexisted. Here, binding energy distributions bifurcated into distinct peaks, separated by a conformational no-man’s-land. Replicas oscillated between states, their transitions governed by energetic barriers and entropic gradients. For FK506, the absence of such transitions—without restraints—highlighted the kinetic traps inherent to large, inflexible ligands.
The analogy to protein folding is instructive. Just as molten globule states mediate folding, partial ligand-protein contacts act as stepping stones toward binding. Accelerating these transitions requires enhanced sampling techniques, but BEDAM’s λ-hopping offers a pragmatic compromise, balancing breadth and depth in conformational exploration.
Ligand Flexibility: A Double-Edged Sword in Drug Design
Flexibility can be a ligand’s ally or adversary. Smaller FKBP12 inhibitors leveraged rotatable bonds to navigate the binding pocket, sampling multiple orientations before settling into the optimal pose. This entropic investment paid dividends, enabling convergence despite sparse structural restraints. FK506, however, demonstrated the pitfalls of rigidity: its cyclic structure hindered adaptive rotations, forcing a “one-shot” binding mechanism reliant on precise preorganization.
The dihedral freedom of ligand sidechains emerged as a critical variable. For compound 2, a single bond rotation bridged unbound and bound states, acting as a conformational switch. FK506’s locked geometry eliminated this pathway, necessitating simultaneous formation of multiple contacts—a rare event under computational timescales. This dichotomy informs drug design: moderate flexibility enhances binding kinetics, but excessive mobility risks entropic penalties.
These insights align with fragment-based drug discovery, where small, flexible probes identify binding hotspots before elaboration. BEDAM’s ability to resolve such nuances positions it as a tool for optimizing “molecular fit,” balancing rigidity and adaptability in lead compounds.
From Simulation to Medicine: Implications for Structure-Based Drug Discovery
The promise of absolute binding free energy methods lies in their versatility. Unlike relative approaches, which compare similar ligands, BEDAM accommodates diverse chemotypes—a boon for scaffold hopping. In FKBP12, the method correctly ranked ligands spanning two orders of magnitude in affinity, despite structural heterogeneity.
Yet, practical challenges persist. The computational cost of unrestrained sampling remains prohibitive for high-throughput screens. Strategic restraints, informed by crystallography or docking, offer a middle ground, prioritizing relevant conformational subspaces. Moreover, integrating BEDAM with experimental data—like cryo-EM or NMR ensembles—could enhance accuracy while preserving dynamic insights.
For drug-resistant targets, where minor mutations distort binding pockets, BEDAM’s capacity to model conformational reorganization is invaluable. By capturing how proteins “breathe” around ligands, it could guide the design of resilient inhibitors, less susceptible to mutational escape.
Future Frontiers: Enhancing Predictive Power in Computational Pharmacology
The path forward demands innovations in both force fields and sampling algorithms. AGBNP2’s shortcomings underscore the need for implicit solvent models that better approximate hydrophobic effects, possibly through machine-learned corrections. Pairing BEDAM with explicit solvent simulations in key regions could hybridize speed and accuracy.
Sampling bottlenecks, exemplified by FK506, call for adaptive λ-scheduling or targeted HREM protocols. Methods like Gaussian Accelerated MD might supplement λ-hopping, accelerating specific degrees of freedom. Additionally, network models mapping conformational landscapes could identify cryptic binding pathways, informing restraint placement.
Lastly, community benchmarking efforts must expand. The FKBP12 study joins a growing corpus validating absolute free energy methods, but diverse systems—membrane proteins, allosteric modulators—will stress-test these tools. As algorithms evolve, so too will our understanding of molecular recognition’s intricate ballet, bringing computational drug design closer to experimental precision.
Study DOI: https://doi.org/10.1021/ct200684b
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Editor-in-Chief, PharmaFEATURES

Subscribe
to get our
LATEST NEWS




Related Posts

Medicinal Chemistry & Pharmacology
Aerogel Pharmaceutics Reimagined: How Chitosan-Based Aerogels and Hybrid Computational Models Are Reshaping Nasal Drug Delivery Systems
Simulating with precision and formulating with insight, the future of pharmacology becomes not just predictive but programmable, one cell at a time.
Medicinal Chemistry & Pharmacology
Coprocessed for Compression: Reengineering Metformin Hydrochloride with Hydroxypropyl Cellulose via Coprecipitation for Direct Compression Enhancement
In manufacturing, minimizing granulation lines, drying tunnels, and multiple milling stages reduces equipment costs, process footprint, and energy consumption.

Medicinal Chemistry & Pharmacology
Decoding Molecular Libraries: Error-Resilient Sequencing Analysis and Multidimensional Pattern Recognition
tagFinder exemplifies the convergence of computational innovation and chemical biology, offering a robust framework to navigate the complexities of DNA-encoded science