The Evolutionary Arms Race in Drug Discovery
The convergence of fragment-based drug design (FBDD) and dynamic combinatorial chemistry (DCC) represents a paradigm shift in medicinal chemistry. FBDD begins with low-affinity molecular fragments, which are iteratively optimized into potent compounds. Traditional methods, however, often involve laborious synthesis and screening. DCC circumvents these challenges by leveraging reversible reactions to generate dynamic libraries (DCLs), where external templates—such as enzymes—shift equilibrium toward high-affinity binders. This adaptive process mimics molecular evolution, enabling the spontaneous emergence of optimized ligands.
By integrating FBDD’s precision with DCC’s efficiency, researchers create a symbiotic platform. Fragments act as seeds, while DCC’s reversible linkages allow structural diversification without stepwise synthesis. This approach eliminates the need for exhaustive purification, as libraries self-select based on biological relevance. The result is a streamlined pipeline where lead compounds evolve in situ, guided by the target’s own binding preferences.
The study’s focus on α-glucosidase inhibitors exemplifies this synergy. Starting with five fragments adhering to the “rule of three”—molecular weight under 300, limited hydrogen bond acceptors, and moderate lipophilicity—researchers constructed acylhydrazone-based DCLs. These reversible linkages enabled rapid exploration of chemical space, with the enzyme itself acting as a template to enrich potent inhibitors.
Diabetes and the Glycemic Tightrope
Diabetes mellitus hinges on dysregulated carbohydrate metabolism, where α-amylase and α-glucosidase catalyze polysaccharide breakdown into glucose. While α-amylase initiates starch digestion, α-glucosidase finalizes it at the intestinal brush border, directly impacting blood glucose levels. Current inhibitors like acarbose target both enzymes but cause gastrointestinal distress due to undigested carbohydrates fermenting in the colon.
Selective α-glucosidase inhibition offers a therapeutic advantage by minimizing systemic enzyme disruption. Unlike α-amylase, which operates upstream, α-glucosidase’s localized activity allows targeted intervention. This specificity reduces off-target effects while maintaining glucose modulation. However, achieving selectivity demands molecular precision, as the enzymes share structural similarities in their catalytic pockets.
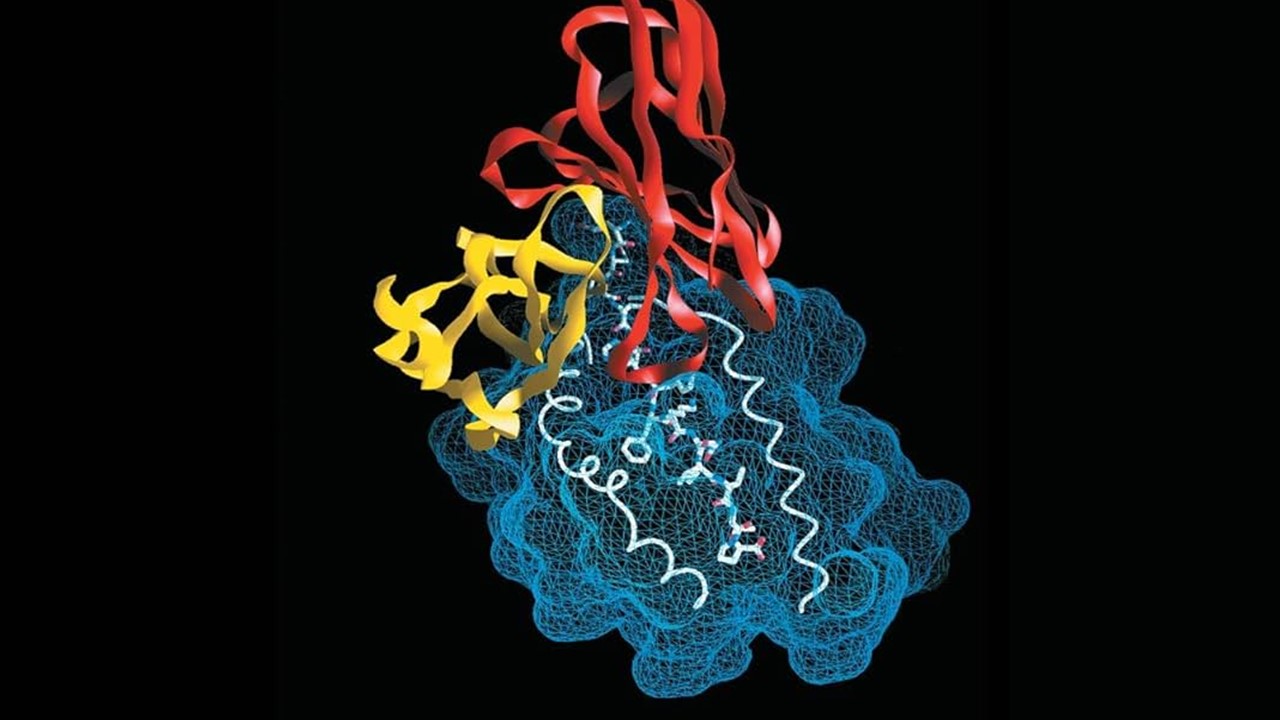
The quest for selective inhibitors drove the design of fragment libraries tailored to α-glucosidase’s active site. Docking studies revealed fragments binding deeply within the catalytic cleft, with a hydrophobic subpocket identified as a growth zone. This region became the focal point for acylhydrazone extensions, designed to exploit π-π stacking and hydrogen bonding with residues like Phe 177 and Arg 312.
Architecting Molecular Libraries with Purpose
Fragment optimization began with constructing DCL-1, combining three acylhydrazides (C1–C3) and five aldehydes (E1–E5). Under physiological conditions, these components reversibly formed 15 acylhydrazones. High-performance liquid chromatography (HPLC) confirmed equilibrium within 16 hours, with α-glucosidase templating amplifying four compounds: C1E3, C2E3, C3E3, and C3E5. Intriguingly, α-amylase failed to perturb the library, hinting at inherent selectivity.
Biological assays revealed C3E5 as the standout, exhibiting moderate inhibition (IC50 ≈32.5 μM) and negligible cytotoxicity. Surprisingly, sulfonate-containing hits like C3E3 showed weak activity, suggesting off-target binding. This underscored the importance of iterative library refinement. DCL-2 emerged as a second-generation library, introducing chromone aldehydes and heterocyclic acylhydrazides to expand hydrophobic and aromatic interactions.
DCL-2’s screening identified C4E5, C4E6, and C4E8 as amplified hits. Among these, C4E8 demonstrated remarkable potency (IC50 ≈2.5 μM), surpassing acarbose and showcasing 80-fold improvement over its parent fragment. Crucially, none inhibited α-amylase, cementing the approach’s selectivity.
From Equilibrium to Elucidation
Kinetic studies illuminated C4E8’s mixed-type inhibition, binding both free enzyme and enzyme-substrate complexes. Lineweaver-Burk plots intersected in the second quadrant, with competitive (Ki ≈1.98 μM) and noncompetitive (αKi ≈2.75 μM) constants indicating dual binding modes. This mechanism allows partial enzyme activity at high substrate concentrations, potentially mitigating toxicity.
Molecular docking visualized C4E8’s binding landscape. The indole moiety engaged Phe 177 and His 239 via π-π stacking, while the chromone’s methyl group nestled into a hydrophobic niche. Hydrogen bonds with Arg 312 and Phe 157 further stabilized the complex, yielding a binding energy of −10.8 kcal/mol. Comparative docking with fragment 5 highlighted the indole extension’s critical role in occupying the target subpocket.
Selectivity as a Therapeutic Imperative
The stark contrast between C4E8’s α-glucosidase inhibition and its inertness toward α-amylase underscores the strategy’s success. Selectivity arises from structural nuances: α-glucosidase’s deeper active site accommodates the indole-chromone scaffold, while α-amylase’s shallower cleft rejects it. This divergence prevents off-target binding, averting gastrointestinal side effects.
Cytotoxicity assessments on HepG2 cells confirmed biocompatibility, with CC50 values exceeding 200 μM for all leads. Such safety profiles position these inhibitors as viable candidates for long-term diabetes management.
A New Paradigm in Enzyme Inhibitor Development
This study exemplifies the power of merging FBDD and DCC. From initial fragments to optimized leads, the process harnesses thermodynamic principles and structural biology to evolve potent, selective inhibitors. C4E8’s success validates the approach, offering a template for targeting other enzymes with precision.
Future directions include in vivo validation and pharmacokinetic profiling. Yet, the methodology’s adaptability—swapping enzyme templates and fragment sets—promises broad applicability. As drug discovery embraces dynamic systems, this hybrid strategy may redefine how we navigate the molecular chess game of therapeutic design.
Study DOI: https://doi.org/10.1021/acsmedchemlett.2c00405
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Editor-in-Chief, PharmaFEATURES

Subscribe
to get our
LATEST NEWS




Related Posts

Medicinal Chemistry & Pharmacology
Aerogel Pharmaceutics Reimagined: How Chitosan-Based Aerogels and Hybrid Computational Models Are Reshaping Nasal Drug Delivery Systems
Simulating with precision and formulating with insight, the future of pharmacology becomes not just predictive but programmable, one cell at a time.
Medicinal Chemistry & Pharmacology
Coprocessed for Compression: Reengineering Metformin Hydrochloride with Hydroxypropyl Cellulose via Coprecipitation for Direct Compression Enhancement
In manufacturing, minimizing granulation lines, drying tunnels, and multiple milling stages reduces equipment costs, process footprint, and energy consumption.

Medicinal Chemistry & Pharmacology
Decoding Molecular Libraries: Error-Resilient Sequencing Analysis and Multidimensional Pattern Recognition
tagFinder exemplifies the convergence of computational innovation and chemical biology, offering a robust framework to navigate the complexities of DNA-encoded science