Drug Product Performance
The origins of which are found to be versatile such as from microbes, minerals, animals, plants, laboratory synthesis, and biotechnological production, a drug is defined as a chemical substance intended for use in the diagnosis, cure, mitigation, treatment, or prevention of disease.

These chemicals are available in an array of pharmaceutical dosage formulations or simply drug products which may be in solid formats as in tablets, lozenges, or capsules, semisolid preparations like creams and ointments, liquid medications as solutions, suspensions or emulsions, and they may be employed for their local therapeutic effects or their systemic activity. Such drug products may even present themselves as drug delivery systems capable of targeted transport of active ingredients to the intended locale of action as in gastric acid-protected or enteric coated tablet formulations or liposomal structures for drug bioavailability improvement. They are especially designed to achieve patient’s specific needs in terms of dosing, maintenance of steady-state concentration, safety, toxicity reduction, palatability and convenience of drug administration.
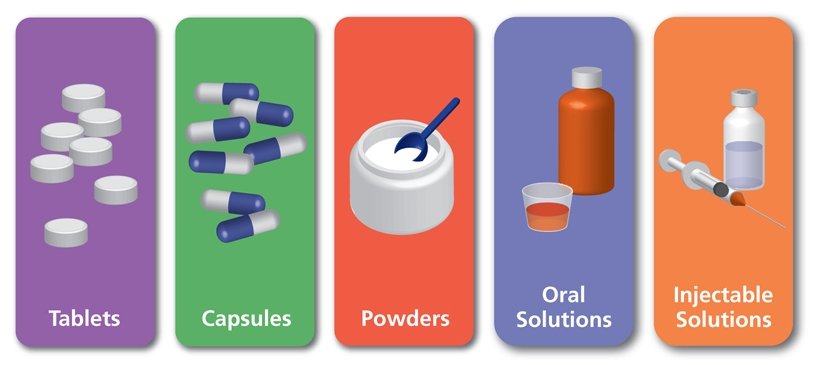
In this regard a drug product performance is best defined to be the release of a biologically active ingredient from the drug product meant either localized drug activity or for plasma absorption to induce systemic effects. Thus, it is imperative and wise that some of the most contemporary cutting-edge pharmaceutical technology and manufacturing endeavors double down on producing drug products of the highest quality with better safety profiles, augmented anti-pathologic effectiveness, and overall significantly more convenience of administration, whether via ingestion, inhalation or injection.
Biopharmaceutics
Biopharmaceutics studies the effects of the physicochemical properties of an active ingredient (drug), the drug product (dosage form) in which the active ingredient is presented as, and the route of administration of the medication on the rate and extent of drug absorption in the systemic circulation. The significance of the drug and its formulation on systemic absorption, and in vivo distribution of the active ingredient to the biological site of action, is identified to be a cascade of activity preceding the manifestation of a drug’s pharmacologic effect. The figure below illustrates the aforementioned dynamic relationship.
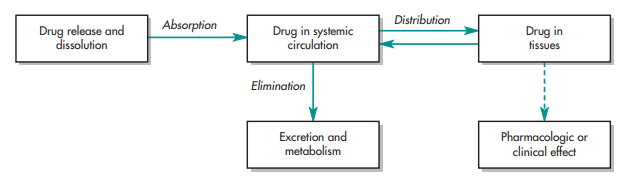
Initially, the active ingredient in its available dosage format is taken by the patient with a dosage presentation-contingent route of administration. Subsequently, the medication is released from its dosage product in a predictable and characterizable manner. Next, a portion of the drug is absorbed from the administration site into either inside the body, at the surrounding tissues, or both. Ultimately, the drug arrives at its intended biological site of action. When the drug concentration at the biological site of action achieves or surpasses the minimum effective concentration (MEC), a therapeutic response is elicited. It is during the clinical trials that the apt dosage regimen, inclusive of initial or bolus dose, maintenance dosages, dosage formats, and interval of drug administration or dosing, is identified. This is to ensure that the medications administered to patient volunteers are not only pharmacologically active but also therapeutically effective. It is good to note that not only do the dosage formulation design and drug’s physical and chemical properties broadly affect the bodily movement of the administered medication but in fact, at times, specially customize the active ingredient’s fate in the body.
A solid combination of physics, chemistry and biology, in and itself, biopharmaceutics provides the scientifically sound foundation for the design and development of drug products. An excellent pharmaceutical scientist will take note that the individual phases in the manufacture of a finished dosage formulation are individually significant factors, themselves, that have high impact on the xenobiotic release from the dosage form and the rate and extent to which a drug becomes available at the site of action, otherwise known as drug bioavailability. Referred to as critical manufacturing variables, these vital stages in the manufacturing process must be heavily taken into account as well. As such, these biopharmaceutic considerations for the design of drug products are presented as follows:
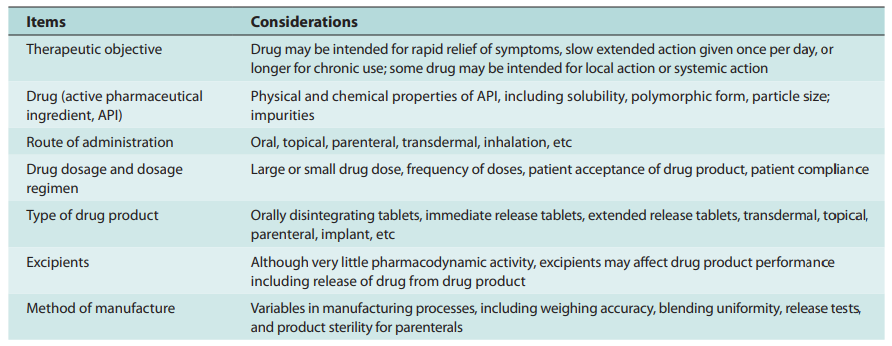
Thus, biopharmaceutics encompasses the necessary understanding and continued research of the factors influencing (1) drug product design, (2) active ingredient stability in dosage formulations, (3) drug dosage form manufacture, (4) biomechanical xenobiotic release from its dosage presentation, (5) dissolution rate, and (6) delivery to its intended site of action either for drug local effects or systemic absorption.
The understanding of these complex relationships is a must for both pharmaceutical scientist and pharmacist in order to determine the most appropriate drug dosage format to contain the active ingredient so as to optimize therapeutic outcomes and reduce unwanted effects.
Biopharmaceutical research endeavors are hinged upon rudimentary principles of science and experimental methodologies. Biopharmaceutical studies utilize in vitro (“in the glass”) and in vivo (“within the living”) methodologies. The non-involvement of laboratory animals or human subjects for experimentation and the sole focus on employment of procedures using only test apparatus and pieces of science equipment are referred to as the former. The latter methodologies are far more complex as they involve animals and/or humans. Together, in vitro and in vivo studies aim to ascertain the influence of a drug’s physiochemical properties, its stability, and the mass manufacture of the drug and its intended dosage formulation on the drug’s biologic performance. Additionally, biopharmaceutics must consider the behavior of the drugs and its dosage format in a physiologic, especially a diseased state, environment, the active drug ingredient’s predetermined therapeutic application, and the route of administration of the medication.

Pharmacokinetics
The subsequent release of the active pharmacological ingredient or drug from its dosage formulation will be followed by an absorptive mechanism whether via the surrounding tissues where the drug was distributed, the body, or both. This observed drug distribution and drug elimination is non-uniform for every patient but is undeniably characterizable with the employment of statistical techniques and mathematical models. Pharmacokinetics is the study of the effects of biological, chemical and physical forces on drug absorption, drug distribution and drug elimination (i.e., metabolism and excretion). Together, drug distribution and elimination are referred to as drug disposition. A necessary requirement for the identification and/or alteration of individual or group dosing regimens is the comprehensive elucidation of a drug’s drug disposition.
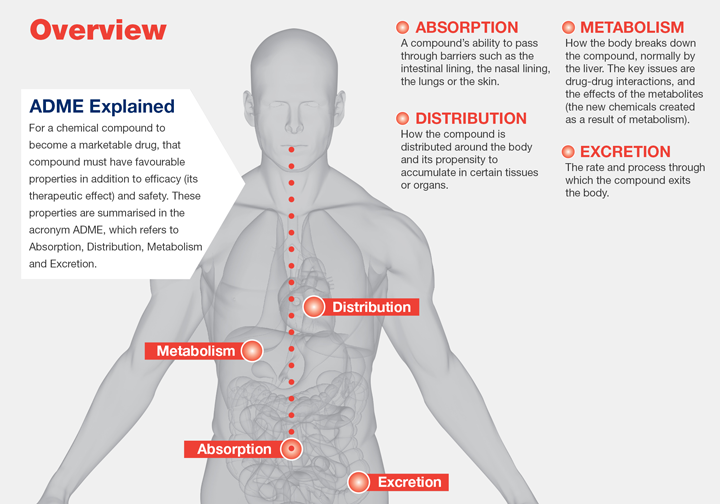
Pharmacokinetic studies encompass both experimental and theoretical approaches. The former focuses on the development of biologic sampling techniques, drug and metabolite measurement using analytical chemistry, and data analytics. The latter aspect anchors on the predictive capabilities of drug disposition models. Hence, the centrality of statistical usage in pharmacokinetic studies cannot be overstated. These statistical methodologies are employed for parametric assessment and data interpretation goaled to design and predict dosing regimens optimized for individuals and for certain patient groups. Statistics proves vital in pinpointing erroneous data and highlighting structural model deviations. The usage of mathematical and computational techniques serve as the core for most methodologies employed in pharmacokinetic studies. The science of theoretical representations revolving around model development and parametrization is referred to as classical pharmacokinetics.
Clinical Pharmacokinetics
A huge number of patients serve as clinical trial volunteers in the process of developing a new drug. This is to ensure that sufficient amount of data regarding the optimum dosage regimens is obtained. These regimens are then recommended in the drug package insert to elicit the intended pharmacologic effect in the majority of the anticipated patient population. Nevertheless, exceptions will always exist as there is no “one dose fits all” in the same way that there is no “single drug fits all”. This is the case with inter- and intraindividual variations causing either subtherapeutic (active drug component concentration below the MEC) or toxic effect (active drug component concentration at or above the minimum toxic concentration, MTC). Adjustment of dosing regimen is, thus, imperative. This is where clinical pharmacokinetics come into play. Clinical pharmacokinetics is the science of applying methodologies employed in pharmacokinetic studies to medication management. By nature, clinical pharmacokinetics is imperatively multifaceted so as to effect individually optimized dosing strategies using the patient’s disease state and full health profile or background.
The study of clinical pharmacokinetics of bioactive drugs in pathologic states necessitates comprehensive inputs from both medical practice and pharmaceutical research. And while it is known that age, gender, genetic variations, and ethnic differences cause pharmacokinetic differences resulting to altered drug therapy outcomes, the influence of diseased states in human cellular and tissue environments on the disposition of the drug is still not well studied. The cornucopia of variations has led to the development of population pharmacokinetics to devote an in-depth understanding of differences in drug pharmacokinetic profiles amongst varying groups of populations.
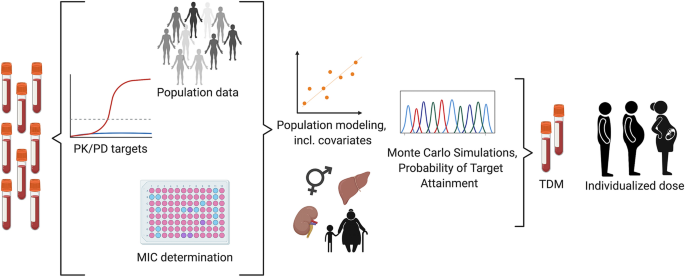
A couple few of drugs have very narrow therapeutic ranges. For these types of highly potent medications, efficacy optimization and adverse toxicity prevention would require the application of clinical pharmacokinetics to therapeutic drug monitoring (TDM). There are two means with which patient monitoring is done with these types of drugs – one is via determination of plasma concentrations of the drug as in the case of the phosphodiesterase inhibiting drug theophylline indicated for COPD and asthma, the other is via monitoring of a specific pharmacologic effect such as prothrombin clotting time as is the case with the vitamin K epoxide reductase inhibitor, anticoagulant warfarin. The clinical pharmacokinetic service (CPKS) ensures safe drug monitoring by providing drug analytics and pharmacokinetic profiling services. Some drugs frequently monitored are the aminoglycosides and anticonvulsants. Since the efficiency of aminoglycosides depends on their presence in sufficient quantities in the blood, it is crucial to keep an eye on their concentration. In addition, aminoglycosides are linked to major toxic side effects, such as acute kidney damage (nephrotoxicity) and damage to hearing and/or balance (ototoxicity). When given intravenously, a number of anticonvulsants have the potential to produce negative cardiac consequences. As a result, patients should be hooked up to a heart monitor and have their vital signs checked often. Unwanted side effects must be reported right away to the doctor. In order to reduce unfavorable side effects, chemotherapy treatments are among other medications that undergo careful monitoring.
Relationship of Drug Concentrations to Drug Response
Drug therapy commencement springboards with the drug manufacturer’s recommended dosage regimen inclusive of the dosage of the bioactive ingredient and the dosing frequency (e.g., 500 mg every 6 hours). Attributable to genetic variations for every patient or their specific pharmacokinetic profile, the dosage regimen provided by the drug manufacturer may not actually elicit the needed expected pharmacologic effect. With the utilization of plasma drug concentrations as bases for clinical effectivity measurement, an objective confirmation can be achieved as to the whether the dosage was found to be subtherapeutic due to the patient’s individual pharmacokinetic profile (verified via low plasma drug concentrations) or the patient was not responsive to the drug therapy at all but this time due to the genetic difference in receptor response. Such cases are to be considered with much scrutiny since the drug concentrations are actually in the therapeutic range but the patient proves to be irresponsive to the initiated drug treatment. On the contrary, some patients are highly responsive the treatment even at lower drug dosages resulting in lower drug concentrations. Other patients aren’t so lucky at all to actually require higher drug concentrations to attain the desired pharmacologic response which would make higher drug doses imperative. The figure below illustrates the concentration of drug in the human body can range from subtherapeutic to toxic.
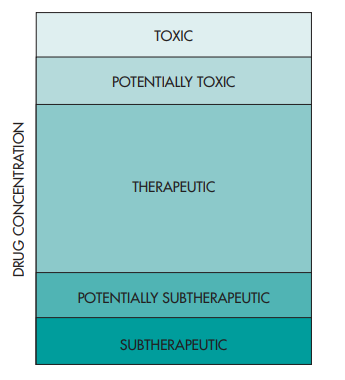
It is preferable, and more often than not observed, that untoward effects to the medications happen during concentrations of the drug at higher levels with respect to the therapeutic drug concentrations. Nevertheless, the case for potent drugs seem to not follow this trend since adverse drug effects are still existent even in therapy-effective drug concentration levels.
Pharmacodynamics
Pharmacodynamics studies the relationship between the bioactive component (drug) concentration at the biological site of action (receptor) and the elicited pharmacologic response, alongside the physiologic effects and biochemical modifications that alter the interaction of the drug with the receptor. The said drug-receptor interaction results in a cascade of biomolecular activities generating a pharmacologic effect – which may be augmentative, inhibitive, neutralizing, or restorative, a toxicologic response, or both. Models of pharmacokinetics and pharmacodynamics are illustrated in such a way that a clear relationship between the plasma drug level and the drug’s bioavailability or its concentration at the biological site of action. These models are made to establish the intensity and time course (onset, duration, residual effects manifestation) of the drug.

Drug Exposure and Drug Response
By simple definition, drug exposure is simply the dose of the drug. An extended characterization for pharmacokinetic profiling purposes includes certain measures of acute or integrated plasma drug concentrations or other biological fluid drug levels namely the Cmax, Cmin, Css, Cmean or Cavg, and the AUC. Cmax is the highest (or peak) blood or serum concentration a medication can reach in a particular body compartment or test region after administration but before the subsequent administration of another dose. The minimum (or trough) concentration that a medication reaches after dosage, or Cmin, is the opposite of Cmax. The time at which the Cmax is noticed is known as the related pharmacokinetic parameter tmax. Since the concentrations are constantly dropping after the dose, the experimental technique has a significant impact on the Cmax and tmax following an intravenous administration. However, upon oral administration, Cmax and tmax depend on the degree, rate, and disposition profile of the medication. They could be used to describe the characteristics of several formulations within the same subject. While the therapeutic benefit of a drug with sustained duration of action typically occurs at concentrations somewhat above the Cmin, short-term drug side effects are most likely to occur at or close to the Cmax. In an effort to demonstrate bioequivalence (BE) between a generic and innovator drug product, the Cmax is frequently assessed. The FDA claims that systemic exposure-related pharmacokinetic parameters like AUC and Cmax are crucial to drug quality bioavailability (BA) and bioequivalence (BE). The area under the curve (AUC), in the discipline of pharmacokinetics, is the definite integral of a drug’s blood plasma concentration as a function of time (this can be done using liquid chromatography–mass spectrometry). In actual practice, the trapezoidal rule is employed to estimate AUC while the drug concentration is monitored at specific discrete points in time. The cumulative drug exposure over time is represented by the AUC (from zero to infinity). When attempting to compare whether two dose forms (for instance, a capsule and a tablet) produce equivalent quantities of tissue or plasma exposure, AUC is a helpful statistic. The therapeutic drug monitoring (TDM) of medications with a restricted therapeutic index is another application. Knowing the average concentration over a period of time, AUC/t, is useful. AUC is sometimes used when discussing elimination. The mass that the body expels is determined by clearance (volume/time) * AUC (mass*time/volume). Steady-state concentration (Css) happens when a drug is administered continuously or repeatedly and the amount being absorbed is the same as the amount being excreted from the body. The period of time when the drug’s concentration in the body remains constant is known as the steady-state concentration. Cavg or Cmean is the average concentration of a drug in the central circulation during a dosing interval in steady state.
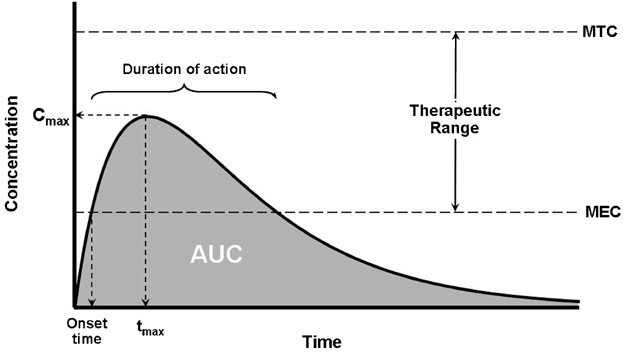
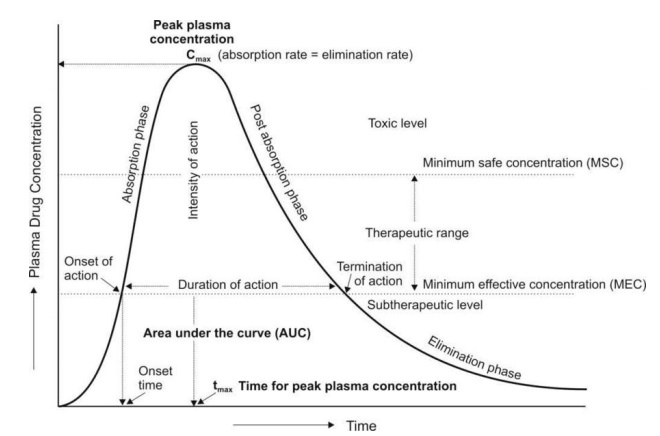
On the other hand, drug response is the direct measure of the pharmacologic effect of the drug. Drug response encompasses a wide range of biomarkers and endpoints ranging from remote biomarkers (like receptor occupancy), to a postulated biomechanistic response (like COX-2 inhibition), to a probable or likely substitute (as in dropping of mean arterial pressure, increased fatty acid beta-oxidation, or reduced cardiac output), and to the complete range of long-term and short-lived clinical effects relevant to either safety or effectiveness.
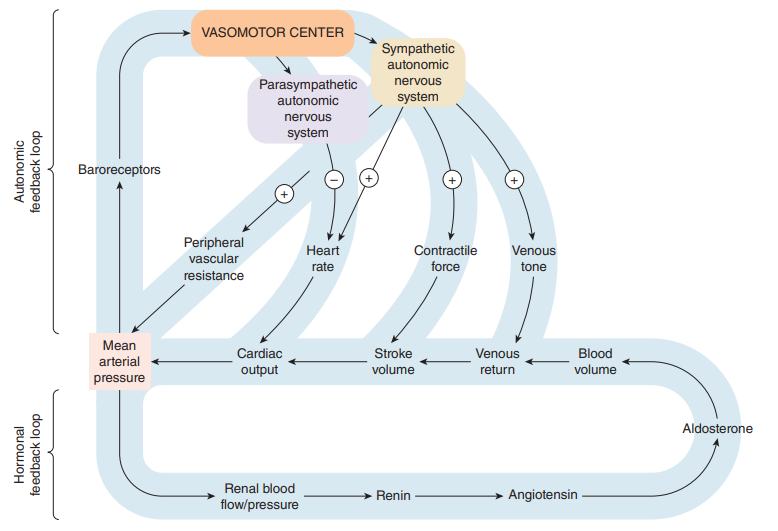
Studies on efficacy and toxicology generate the required information on the effectiveness and safety profiles of the active drug ingredient or drug during its development. These data are particularly important for assessment of drug effectiveness and safety in patients with hepatic concerns and renal insufficiencies, subjects referred to as special patient populations. The clinical utility of most medications is hinged upon gauging the benefits of favorable outcomes against the risks caused by the unfavorable ones at certain doses. An even special medications category namely the potent drugs, dosages and rate of administration make it particularly imperative for titrations to be set in place so as to attain desired pharmacologic effects and to improve tolerance to adverse drug reactions.
Toxicokinetics and Clinical Toxicology
The application of principles of pharmacokinetics to the design, conduct, and interpretation of drug safety evaluation studies is referred to as toxicokinetics. This field of study also validates dose-related exposure in non-human subjects or animals. Toxicokinetic data provide great assistance in the exegesis of findings about the toxicologic profiles in animal subjects and the extrapolation of the animal data interpretations to humans. It is during the preclinical drug development phase or the exploratory phase when the toxicokinetic studies are performed in animals. These studies have high expectations of being continued even after the drug candidate has been subjected in clinical trials.
Clinical toxicology is the science of toxicants (poisons) and drug’s adverse effects in the human body. According to Paracelsus, the only difference between a drug and a poison is the dose. In an overdosed individual, the drug’s pharmacokinetics are likely to be significantly different from the same drug’s pharmacokinetics at subtherapeutic or lower therapeutic doses. Since pharmacologic effects are mainly mediated via receptor-drug interactions, a drug in very high doses is expected to saturate enzymes that participate in the absorption, biotransformation or metabolism, or active renal secretion mechanisms of the body. Subsequently, the pharmacokinetics of the drug is transformed from a linear type to a non-linear one. We anticipate that as the dose of a drug is increased, the steady-state concentration will rise correspondingly. For example, if the dosing rate is doubled or halved, the plasma drug concentration will likewise double or halve, respectively. The plasma drug concentration does, however, occasionally vary more or less than would be predicted from a change in dose rate. This is referred to as non-linear pharmacokinetic behavior, which might be problematic when changing doses. Medications typically associated with cases of overdosage, hence of toxicity, include tricyclic antidepressants (TCAs), morphine, salicylates, and acetaminophen or paracetamol. Fortunately, several of these drugs are easily assayed using fluorescence immunoassay (FIA) kits.
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Editor-in-Chief, PharmaFEATURES

Subscribe
to get our
LATEST NEWS




Related Posts

Medicinal Chemistry & Pharmacology
Invisible Couriers: How Lab-on-Chip Technologies Are Rewriting the Future of Disease Diagnosis
The shift from benchtop Western blots to on-chip, real-time protein detection represents more than just technical progress—it is a shift in epistemology.

Medicinal Chemistry & Pharmacology
Designing Better Sugar Stoppers: Engineering Selective α-Glucosidase Inhibitors via Fragment-Based Dynamic Chemistry
One of the most pressing challenges in anti-diabetic therapy is reducing the unpleasant and often debilitating gastrointestinal side effects that accompany α-amylase inhibition.

Medicinal Chemistry & Pharmacology
Into the Genomic Unknown: The Hunt for Drug Targets in the Human Proteome’s Blind Spots
The proteomic darkness is not empty. It is rich with uncharacterized function, latent therapeutic potential, and untapped biological narratives.

Medicinal Chemistry & Pharmacology
Aerogel Pharmaceutics Reimagined: How Chitosan-Based Aerogels and Hybrid Computational Models Are Reshaping Nasal Drug Delivery Systems
Simulating with precision and formulating with insight, the future of pharmacology becomes not just predictive but programmable, one cell at a time.
Read More Articles







Drug Discovery Biology
April 17, 2025
Myosin’s Molecular Toggle: How Dimerization of the Globular Tail Domain Controls the Motor Function of Myo5a
Myo5a exists in either an inhibited, triangulated rest or an extended, motile activation, each conformation dictated by the interplay between the GTD and its surroundings.

{kind=link}