In the bygone era, drug discovery was akin to a blindfolded journey through a labyrinth, relying solely on trial and error. Anything remotely promising was subjected to clinical experimentation to assess its efficacy. However, the landscape changed with the dawn of chemistry, where researchers began isolating active compounds from medicinal plants, delving deeper into their molecular structures. As molecular biology and biochemistry stepped into the limelight, elucidating the intricate dance between genes and ligands, the foundation for modern drug design was laid. The pinnacle of this evolution was marked by the advent of genomics, proteomics, bioinformatics, and chemoinformatics, ushering in an era of large-scale screening and in silico experimentation.
Deciphering Pharmacophore Models
At the heart of drug design lies the pharmacophore – a molecular blueprint embodying the essential features crucial for a drug’s biological activity. Through the lens of a pharmacophore model, researchers peer into the intricate interplay between key amino acids nestled within the active site of a target protein. By scrutinizing binding sites and potential interactions, pharmacophore models unveil the molecular landscape governing ligand-protein interactions. These models, derived from known ligands or protein structures, serve as guiding beacons in the quest for novel therapeutics, particularly in designing specific inhibitors for disease-related proteins like G-protein coupled receptors and enzymes.
Crafting the Pharmacophore Blueprint
Crafting a pharmacophore model is no trivial feat, requiring a meticulous orchestration of ligand selection, conformation generation, and feature identification. Ligands, sourced from databases or literature, undergo stringent scrutiny to ensure uniformity in biological mechanisms and structural integrity. As the backbone of the model takes shape, descriptors are meticulously calculated, capturing nuances of molecular structure crucial for biological activity. Through a symphony of alignment algorithms and validation protocols, pharmacophore models emerge as potent tools, capable of illuminating the intricate dance between ligands and their protein counterparts.
Pharmacophore Models in Action
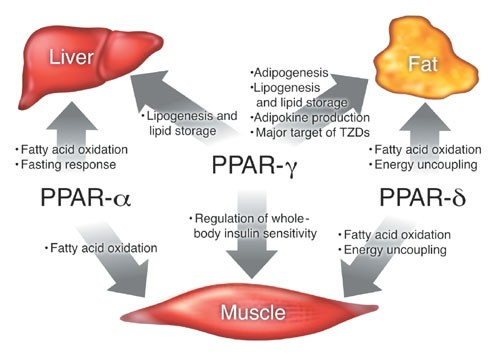
The utility of pharmacophore models transcends theoretical realms, finding practical applications in virtual screening and drug discovery. From unraveling Myc-Max heterodimer disruptors to identifying peroxisome proliferator-activated receptor-γ (PPAR-γ) partial agonists, pharmacophore models serve as compasses, guiding researchers through the labyrinth of drug discovery. Whether as precursors to virtual screening or as filters to sift through molecular databases, these models offer invaluable insights into the design and optimization of therapeutics, heralding a new era of precision medicine.
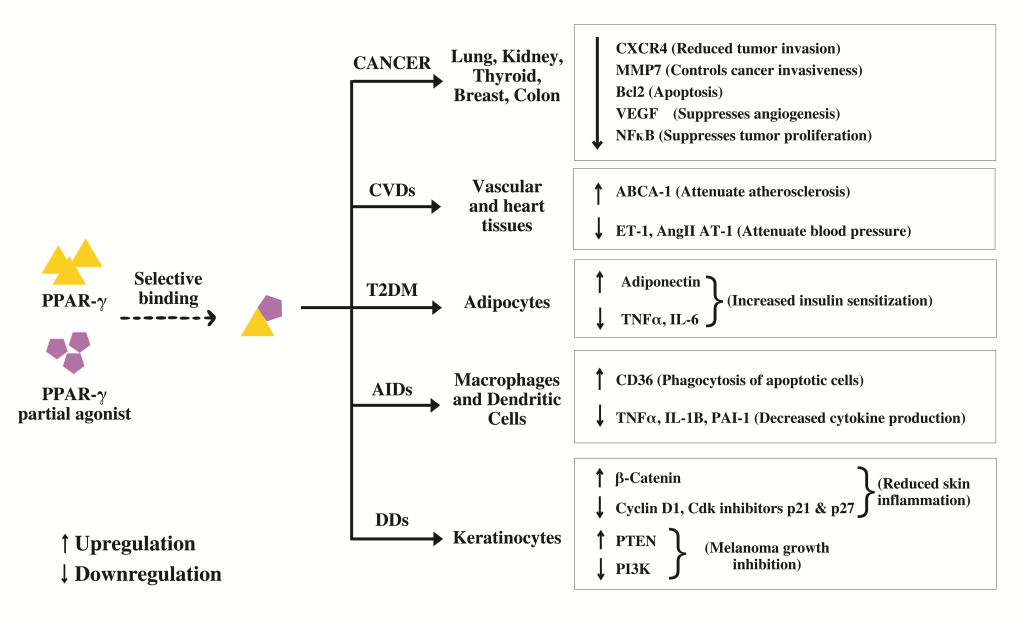
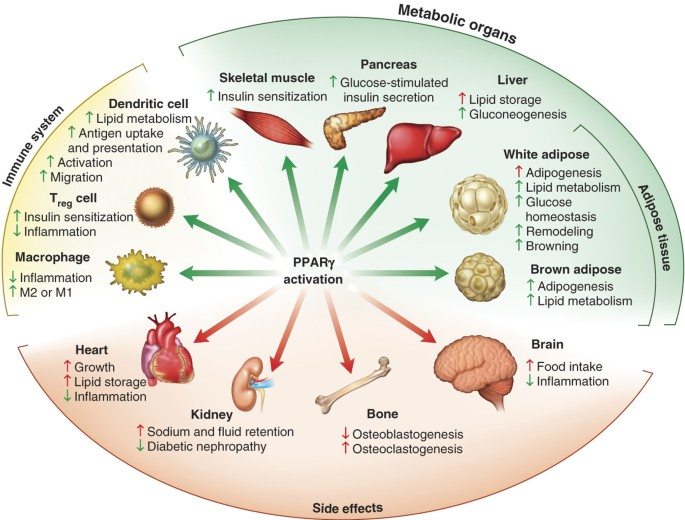
QSAR: Chemical Biology on Roids
Quantitative Structure-Activity Relationships (QSAR) stands as a cornerstone in the arsenal of rational drug design, offering a quantitative lens into the interplay between chemical structure and biological activity. Unlike pharmacophore models, QSAR delves into the quantitative realm, unraveling the nuanced effects of chemical properties on ligand activity. Through a meticulous dance of descriptor selection and modeling methods, QSAR unveils the intricate web of molecular interactions, paving the way for predictive modeling and virtual screening.
Constructing the QSAR Scaffold
Building a QSAR model entails a meticulous choreography of ligand selection, descriptor calculation, and modeling method selection. From the gathering of ligands and activity data to the calculation of structural descriptors, each step in the QSAR pipeline demands precision and attention to detail. Through a symphony of statistical algorithms and validation techniques, QSAR models emerge as predictive powerhouses, capable of unraveling the intricate relationship between chemical structure and biological activity.

Harnessing the QSAR Potential
From predicting the activity of novel compounds to unraveling the mysteries of drug-induced cardiotoxicity, QSAR finds myriad applications across diverse domains. Whether in deciphering the potency of CCR2 inhibitors or assessing drug delivery efficiency in eye drop medications, QSAR serves as a versatile tool in the arsenal of drug design. By leveraging QSAR models as gateways to predictive modeling and virtual screening, researchers embark on a journey of discovery, unveiling novel therapeutics with unprecedented precision.
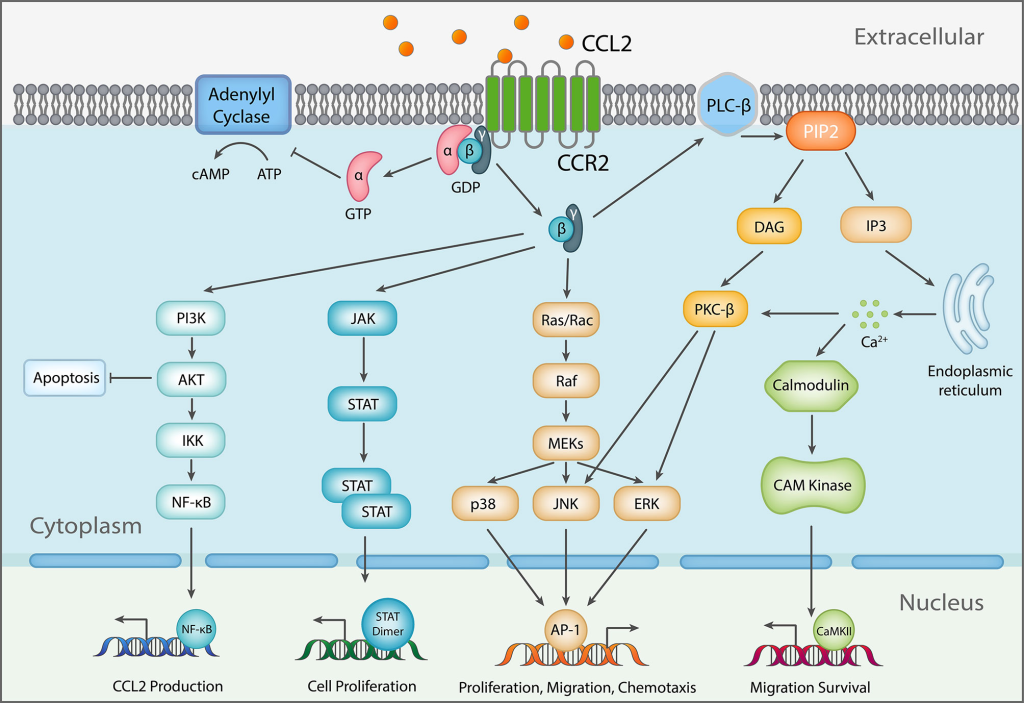
Pharmacophore Models of ATP Synthase Beta Subunit-Binding Ligands
Pharmacophore models illuminate the path to novel oncotherapeutic drug design, particularly in targeting ATP synthase beta subunits. Aurovertin B, known for its affinity to the beta subunits of ATP synthase, shows promise as a potential breast cancer therapy, However, due to its complex structure, synthesizing Aurovertin B poses significant challenges, necessitating the development of new ATP synthase-targeting drugs for cancer research.

To achieve this, it’s crucial to identify the common backbone structure and essential functional groups shared by ligands. While aligning all molecules may reveal a common backbone, critical functional groups might not be evident. Hence, a pharmacophore model was constructed to identify these shared functional groups. Once identified, the common backbone structure can be deduced by eliminating branches lacking necessary functional groups, provided the molecules align well with the pharmacophore and each other.
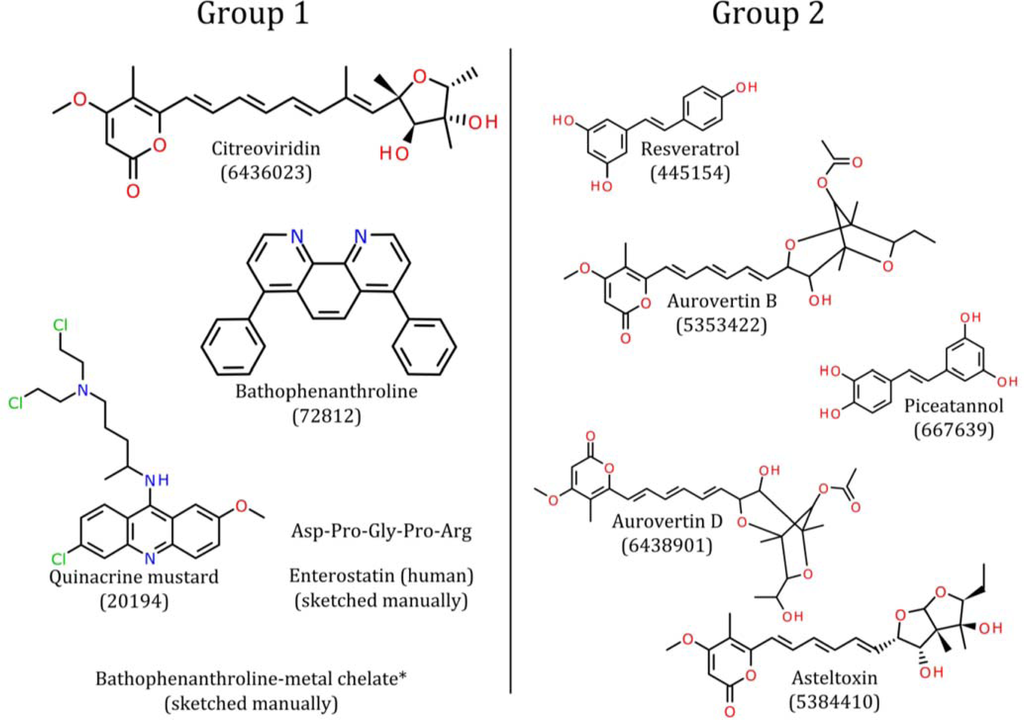
By dissecting the molecular landscape of known ligands and unraveling common functional groups, pharmacophore models offer glimpses into the intricate dance between ligands and their protein targets. Through meticulous data curation and algorithmic wizardry, these models unveil the structural blueprint for designing next-generation inhibitors, heralding a new dawn in cancer research.
Drug Discovery, Brighter than Ever
As we traverse the frontiers of rational drug design, the synergy between QSAR and pharmacophore modeling emerges as a beacon of hope. From the unraveling of molecular mysteries to the design of precision therapeutics, these techniques offer a glimpse into the future of drug discovery. Armed with a deep understanding of their principles and applications, researchers stand poised to unlock the secrets of the molecular world, ushering in a new era of precision medicine and transformative healthcare paradigms.
Study DOI: 10.3390%2Fijms12085304
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Editor-in-Chief, PharmaFEATURES

Subscribe
to get our
LATEST NEWS
Related Posts

Drug Discovery Biology
Proteolytic Rewriting: Engineering Controlled Absence of Pathogenic Protein Persistence
Targeted protein degradation transforms drug therapy by engineering the cellular machinery to erase, rather than merely inhibit, pathogenic proteins.

Drug Discovery Biology
Open Nucleotides: Grant-Driven Infrastructure for Equitable mRNA Vaccine Manufacturing
By developing accessible cap analogs and RNA raw materials, Hongene Biotech, guided by David Butler’s expertise in nucleotide chemistry and supported by the Gates Foundation, is reshaping the molecular infrastructure that underpins global mRNA vaccine equity.
Drug Discovery Biology
Redox Messengers: How Endogenous Gasotransmitters Rewire Vascular Biology to Resist Age-Driven Oxidative Stress
Gasotransmitters provide a biologically sophisticated means of counteracting age-related oxidative stress and preserving vascular resilience.
Drug Discovery Biology
Aptamer Signal Dynamics: Engineering Nucleic Acid Recognition Systems for High-Fidelity, Multi-Modal Biosensing
Aptamers redefine biosensing by pairing programmable molecular recognition with versatile transduction strategies capable of detecting clinically relevant biomarkers with exceptional fidelity.
Read More Articles
Medicinal Chemistry & Pharmacology
March 6, 2026
Algorithmic Therapeutics: Artificial Intelligence Reshaping Biotech Drug Discovery and Product Development
Artificial intelligence is transforming biotech by making therapeutic discovery less like screening for luck and more like engineering across molecules, biologics, and delivery systems.


Chemistry, Manufacturing & Controls
March 4, 2026
Factory Logic: Modular Pharmaceutical Manufacturing Enabling Rapid Drug Production and Continuous Innovation
Modular pharmaceutical factories transform drug manufacturing into a continuously evolving technological system capable of integrating new therapies, processes, and production innovations without disrupting regulated operations.



Medicinal Chemistry & Pharmacology
February 16, 2026
Spatial Collapse: Pharmacologic Degradation of PDEδ to Disrupt Oncogenic KRAS Membrane Localization
PDEδ degradation disrupts KRAS membrane localization to collapse oncogenic signaling through spatial pharmacology rather than direct enzymatic inhibition.

Medicinal Chemistry & Pharmacology
February 4, 2026
Neumedics’ Integrated Innovation Model: Dr. Mark Nelson on Translating Drug Discovery into API Synthesis
Dr. Mark Nelson of Neumedics outlines how integrating medicinal chemistry with scalable API synthesis from the earliest design stages defines the next evolution of pharmaceutical development.

Clinical Operations
February 3, 2026
Zentalis Pharmaceuticals’ Clinical Strategy Architecture: Dr. Stalder on Data Foresight and Oncology Execution
Dr. Joseph Stalder of Zentalis Pharmaceuticals examines how predictive data integration and disciplined program governance are redefining the future of late-stage oncology development.