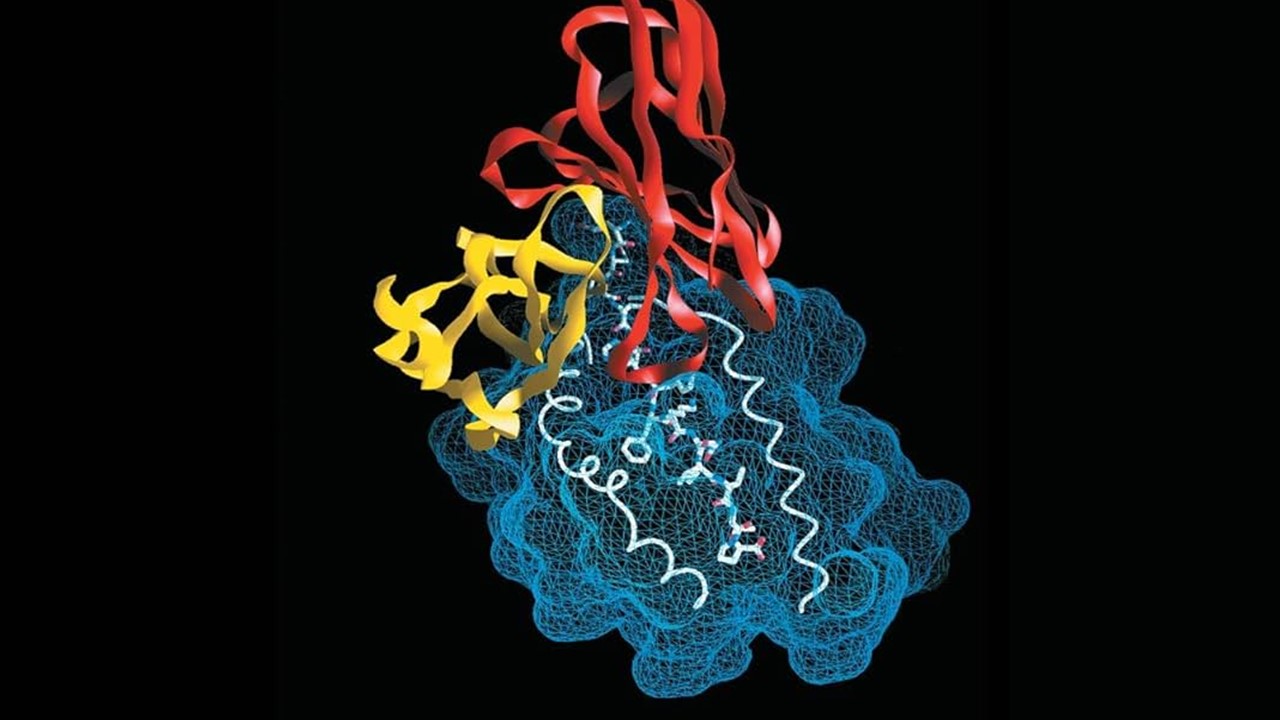
The Evolution of AI in Molecular Design
The pharmaceutical industry has long been defined by its rigorous and costly approach to drug discovery. Traditional methods, reliant on high-throughput screening and expert-driven molecular synthesis, have succeeded in bringing effective drugs to market but at an immense financial and temporal cost. The sheer complexity of chemical space, estimated to encompass anywhere between and synthetically feasible molecules, makes the search for novel therapeutics akin to finding a needle in an astronomical haystack. In silico methods, particularly those leveraging machine learning, have emerged as transformative tools in this domain, significantly enhancing the efficiency and accuracy of molecular design. However, despite the promises of artificial intelligence (AI), existing deep learning approaches struggle with key challenges such as structural repetition, lack of chemical diversity, and difficulties in adversarial training stability.
One of the most promising breakthroughs in AI-driven molecular generation is the development of Reinforced Adversarial Neural Computers (RANC), a deep neural network (DNN) architecture designed to address the inherent limitations of previous generative models. Unlike earlier methods that rely on conventional generative adversarial networks (GANs), RANC integrates a Differentiable Neural Computer (DNC), an architecture equipped with an external memory bank that enhances its ability to retain, manipulate, and generate complex molecular sequences. This additional memory capacity allows RANC to create diverse and chemically valid molecular structures, overcoming the common pitfalls of generative collapse seen in conventional GAN-based frameworks. By refining the balance between generator and discriminator models, RANC not only improves molecular novelty but also preserves essential drug-likeness properties, making it a formidable tool in the next generation of computational drug design.
The significance of this advancement cannot be overstated. AI-driven drug discovery has historically suffered from the inability to reliably generate complex and viable molecular structures that align with real-world medicinal chemistry constraints. Many deep learning models produce molecules that either lack structural relevance or fail to adhere to essential pharmacokinetic properties, such as solubility, bioavailability, and stability. RANC represents a decisive shift by mitigating these challenges through its reinforcement learning framework, ensuring that the generated molecular structures do not merely satisfy arbitrary mathematical objectives but are also chemically meaningful. By bridging the gap between computational efficiency and medicinal applicability, RANC stands as a pivotal innovation in the evolving landscape of AI-enhanced molecular discovery.
Overcoming Adversarial Training Challenges in Molecular Generation
One of the most persistent problems in deep learning-based molecular design is the instability of adversarial training. Conventional GAN-based models, such as the widely recognized ORGANIC (Objective-Reinforced Generative Adversarial Network for Inverse-design Chemistry), utilize a generator-discriminator paradigm in which the generator proposes molecular structures and the discriminator assesses their validity based on reference data. While this system holds promise, it frequently encounters training instabilities that degrade the quality of the generated molecules. Two major pitfalls dominate the field: mode collapse, where the generator produces highly repetitive structures, and the perfect discriminator problem, where the discriminator becomes too powerful, leading to stagnation in generator learning. These issues significantly hinder the ability to create diverse and viable molecular candidates for drug development.
RANC fundamentally redefines this dynamic by integrating a memory-enhanced generator through the use of a Differentiable Neural Computer. The inclusion of an external memory bank allows RANC to retain and recall complex molecular features across training cycles, ensuring that generated molecules are not only structurally diverse but also chemically meaningful. This approach is particularly effective in mitigating the perfect discriminator problem, as the generator can reference past successful molecular patterns and adjust its output accordingly. Unlike ORGANIC, which tends to favor shorter and simpler molecular structures due to adversarial training biases, RANC maintains a balanced distribution of molecular properties, closely resembling those found in medicinal chemistry datasets. By preserving molecular complexity while improving training stability, RANC offers a robust alternative to traditional GAN-based molecular design systems.
The effectiveness of RANC in overcoming adversarial challenges is evident in its ability to generate unique and pharmacologically relevant molecules. Unlike earlier models, which struggle with overly simplistic or repetitive molecular outputs, RANC produces a wide range of novel structures that align with established medicinal chemistry principles. This breakthrough significantly enhances the applicability of AI-generated molecules in real-world drug development scenarios, offering pharmaceutical researchers a tool that is not only computationally efficient but also chemically reliable. By stabilizing adversarial training and reinforcing structural diversity, RANC sets a new benchmark in the application of deep learning for molecular innovation.
Generating Molecular Structures with Enhanced Chemical Fidelity
A major limitation of conventional AI-driven molecular design models is their inability to consistently generate molecules that adhere to fundamental medicinal chemistry principles. While GAN-based models like ORGANIC have demonstrated some success in producing valid molecular structures, they frequently deviate from desired chemical properties such as molecular weight (MW), lipophilicity (logP), and topological polar surface area (TPSA). These properties are critical in drug discovery, as they directly influence a molecule’s absorption, distribution, metabolism, and excretion (ADME) characteristics. A model that fails to preserve these attributes generates compounds that may appear valid on paper but are ultimately unsuitable for pharmaceutical development. The inability to align generated molecular structures with known medicinal chemistry constraints remains a significant challenge in AI-driven drug design.
RANC directly addresses this issue by incorporating reinforcement learning (RL) objectives that prioritize molecular characteristics aligned with drug-likeness criteria. Unlike ORGANIC, which often generates molecules that are structurally dissimilar to the reference dataset, RANC maintains distributions that closely match the training data’s key chemical descriptors. This is particularly evident in the model’s ability to retain appropriate SMILES string lengths, ensuring that generated molecules do not excessively diverge from real-world chemical structures. The inclusion of medicinal chemistry filters (MCFs) further refines the generated molecules, eliminating undesirable substructures such as reactive functional groups, metabolically unstable moieties, and toxic fragments. By rigorously filtering out non-viable candidates, RANC significantly enhances the overall quality and feasibility of AI-generated drug candidates.
Experimental comparisons between RANC and ORGANIC reveal that RANC consistently outperforms its predecessor across multiple evaluation metrics. The model generates a significantly higher proportion of molecules that successfully pass MCFs, demonstrating its superior ability to produce chemically viable structures. Furthermore, RANC-generated molecules exhibit enhanced heterocyclic diversity, an essential feature for optimizing pharmacodynamic and pharmacokinetic properties in drug candidates. By integrating a chemically informed reinforcement learning strategy, RANC ensures that its molecular outputs are not just statistically optimized but also medicinally relevant. This positions RANC as a pivotal advancement in AI-driven molecular design, offering pharmaceutical researchers a more reliable and chemically attuned approach to de novo drug discovery.
A New Era in AI-Driven Drug Discovery
The introduction of RANC marks a significant leap forward in the application of artificial intelligence for drug discovery. While previous models such as ORGANIC have laid the groundwork for AI-driven molecular generation, they remain limited by inherent training instabilities and a tendency to produce molecular structures that lack real-world applicability. RANC overcomes these barriers by leveraging a Differentiable Neural Computer to enhance generator performance, stabilize adversarial training, and ensure that generated molecules retain crucial medicinal chemistry properties. This innovation provides pharmaceutical researchers with a powerful tool capable of exploring vast chemical spaces while maintaining the structural complexity and diversity necessary for effective drug development.
The implications of this advancement extend beyond molecular generation. By refining AI-driven drug design methodologies, RANC has the potential to significantly reduce the time and cost associated with early-stage drug discovery. Traditional pharmaceutical research cycles can take over a decade and require billions of dollars in investment, often with low success rates. AI models like RANC offer an opportunity to streamline this process by identifying promising drug candidates with higher efficiency and accuracy. The ability to generate structurally diverse and medicinally relevant molecules at scale represents a transformative shift in how pharmaceutical research is conducted, bridging the gap between computational prediction and experimental validation.
Moving forward, the continued refinement of AI-driven molecular generation models will be crucial in unlocking the full potential of artificial intelligence in drug discovery. Future research could focus on integrating additional pharmacological constraints, incorporating target-specific activity predictions, and enhancing the interpretability of AI-generated molecular structures. The introduction of quantum chemistry methods, coupled with AI-driven predictive modeling, could further optimize molecular generation for specific therapeutic targets. As AI continues to evolve, models like RANC will play an increasingly integral role in revolutionizing the pharmaceutical industry, ushering in a new era of intelligent drug design that is faster, more efficient, and more precise than ever before.
Study DOI: https://pubs.acs.org/doi/abs/10.1021/acs.jcim.7b00690
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Editor-in-Chief, PharmaFEATURES

Subscribe
to get our
LATEST NEWS




Related Posts

Medicinal Chemistry & Pharmacology
Aerogel Pharmaceutics Reimagined: How Chitosan-Based Aerogels and Hybrid Computational Models Are Reshaping Nasal Drug Delivery Systems
Simulating with precision and formulating with insight, the future of pharmacology becomes not just predictive but programmable, one cell at a time.
Medicinal Chemistry & Pharmacology
Coprocessed for Compression: Reengineering Metformin Hydrochloride with Hydroxypropyl Cellulose via Coprecipitation for Direct Compression Enhancement
In manufacturing, minimizing granulation lines, drying tunnels, and multiple milling stages reduces equipment costs, process footprint, and energy consumption.

Medicinal Chemistry & Pharmacology
Decoding Molecular Libraries: Error-Resilient Sequencing Analysis and Multidimensional Pattern Recognition
tagFinder exemplifies the convergence of computational innovation and chemical biology, offering a robust framework to navigate the complexities of DNA-encoded science