High-Throughput Screening: The New Frontier
High-throughput screening (HTS) has revolutionized the drug discovery landscape, allowing researchers to test thousands to millions of biological, genetic, chemical, or pharmacological samples swiftly. This method accelerates the identification of promising candidates for further study. Integral to this process is the use of assays, which examine substances to determine their purity and properties. The signal detection system of these assays, known as readouts, measures the cell response, thereby providing crucial data about the results.
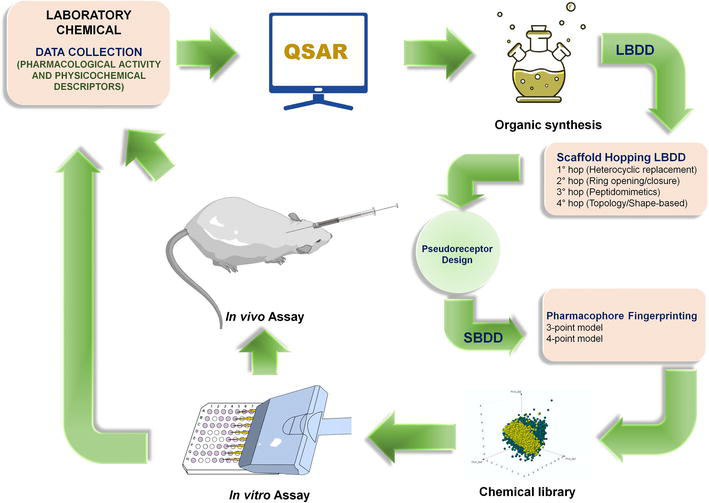
Harnessing Computer-Aided Drug Design
The advent of computer-aided drug design (CADD) marked a significant leap in drug development. This approach leverages various computational techniques supported by both the pharmaceutical industry and academic institutions to expedite the creation of new drugs.
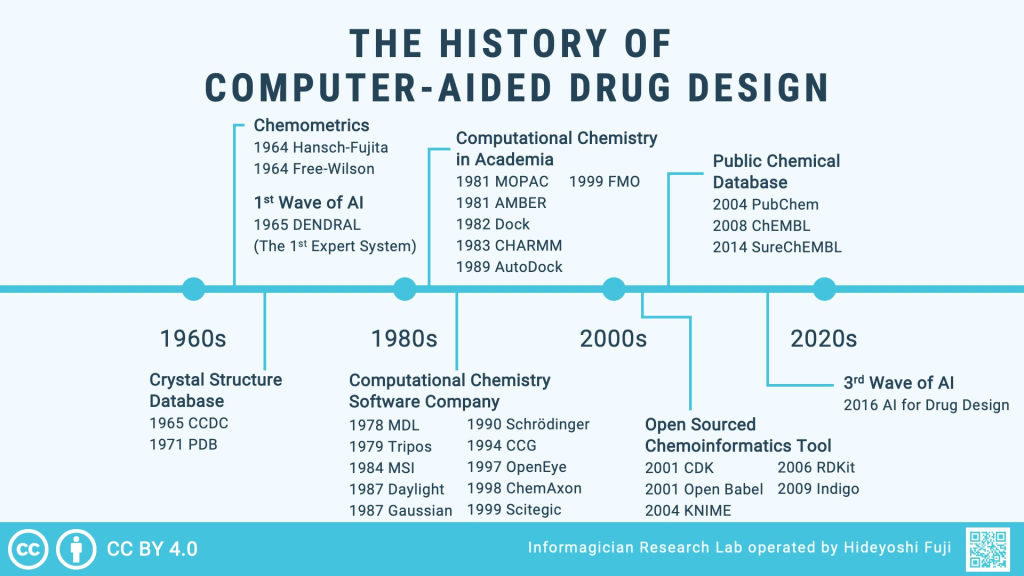
Among these techniques are quantitative structure-activity relationship (QSAR) models, pharmacophore modeling, lead optimization, molecular dynamics, and molecular docking. Recently, machine learning applications have been integrated into these methods, enhancing their efficiency and accuracy.
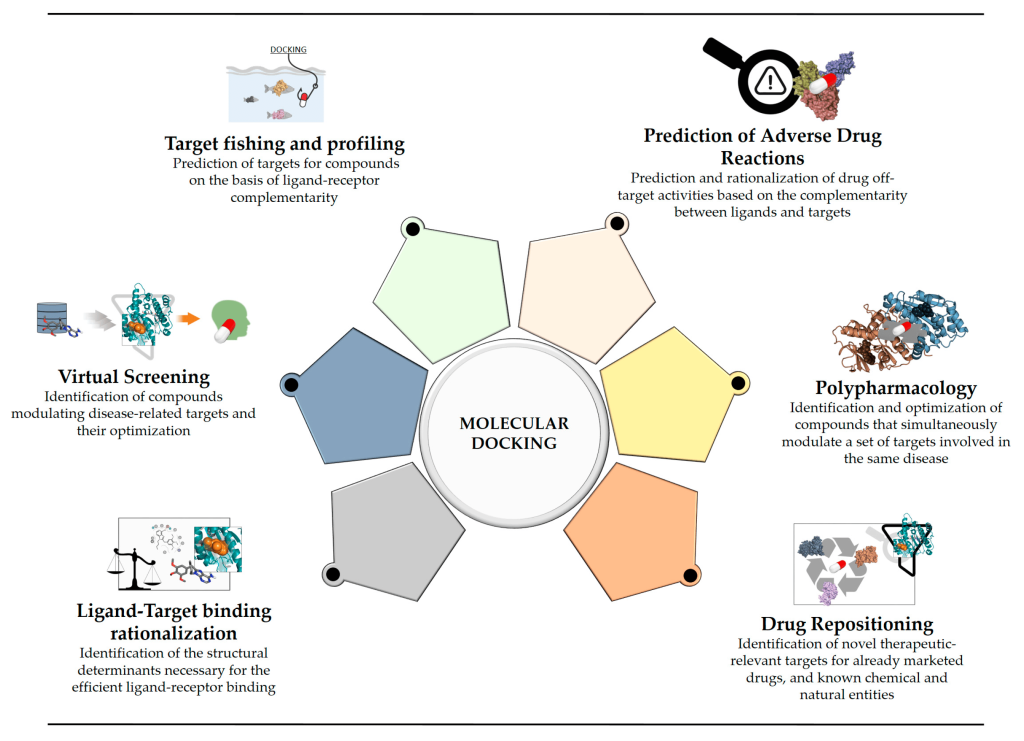
Understanding Molecular Docking
Molecular docking is a computational technique that examines the interactions between a macromolecule, typically a protein, and a ligand. This method, which dates back to the 1980s, began with the molecular modeling of proteins and has since evolved into a cornerstone of modern drug discovery. By evaluating the interaction energy between a target protein and numerous ligands, researchers can identify potential drug candidates. The increase in computational power and the availability of extensive databases of ligands and proteins have made molecular docking indispensable in the search for new drugs.
Decoding the Conformational Search Algorithm
At the heart of molecular docking are two essential components: the conformational search algorithm and the scoring function. The conformational search algorithm explores the conformational space of the ligand at the binding site. There are two primary types of molecular docking: blind docking and binding-site docking. Blind docking involves the entire protein within the sampling space, ideal for identifying the site with the highest affinity energy when the binding site is unknown. In contrast, binding-site docking focuses on a specific area, typically the protein-binding site, or an allosteric site of interest.
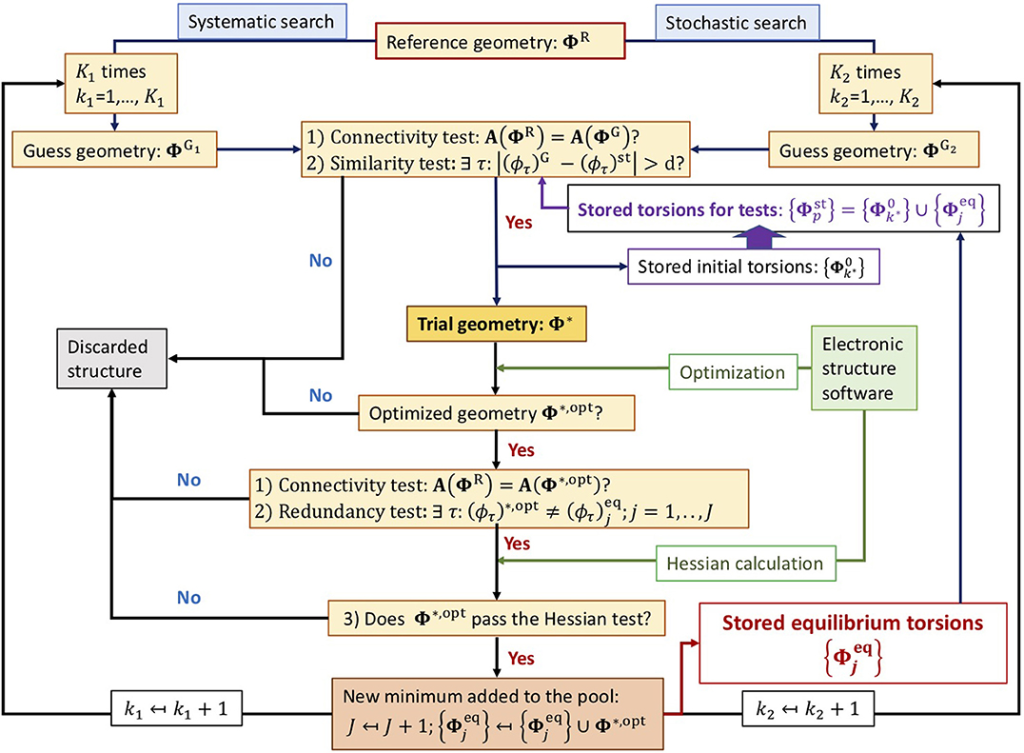
The goal of the conformational search is to generate all possible conformations of the ligand for evaluation. This process considers the ligand’s structural parameters, such as torsion and translation, and its degrees of freedom. The number of conformations increases exponentially with the degrees of freedom, a phenomenon known as combinatorial explosion. Researchers employ either systematic or stochastic methods to perform the conformational search. Systematic methods evaluate small conformational changes until they converge to an energy minimum, while stochastic methods generate random conformations, covering a larger sampling area and increasing the likelihood of finding the global energy minimum.
Scoring Functions: Measuring Interaction Energy
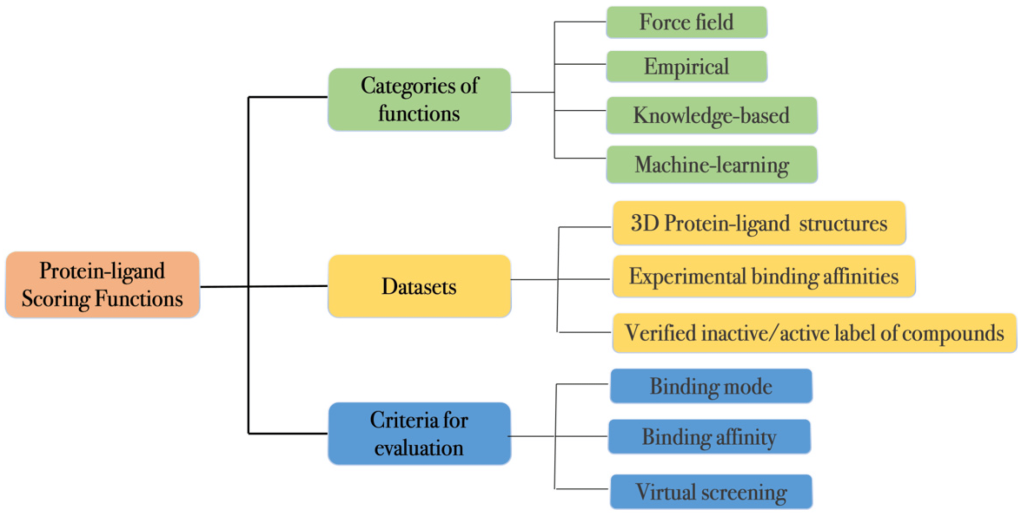
The scoring function is a predictive model that calculates the binding free energy of each ligand-protein conformation. Traditional scoring functions are based on physical calculations of atomic interactions, known as force fields, which include potential energy, torsion terms, bond geometry, electrostatic terms, and the Lennard-Jones potential. Empirical scoring functions use weighted energy terms derived from experimental data, including hydrogen bonding, van der Waals interactions, and hydrophobicity. Knowledge-based scoring functions, on the other hand, rely on statistical models derived from large databases of ligand-protein complexes.
Validating molecular docking protocols involves comparing the docking results with crystallography data using the root mean square deviation (RMSD). The DockBench platform facilitates this validation by performing autocoupling routines to replicate crystallographic complexes, thus measuring the ability of each protocol to reproduce the crystallographic pose.

Consensus Docking: A Multipronged Approach
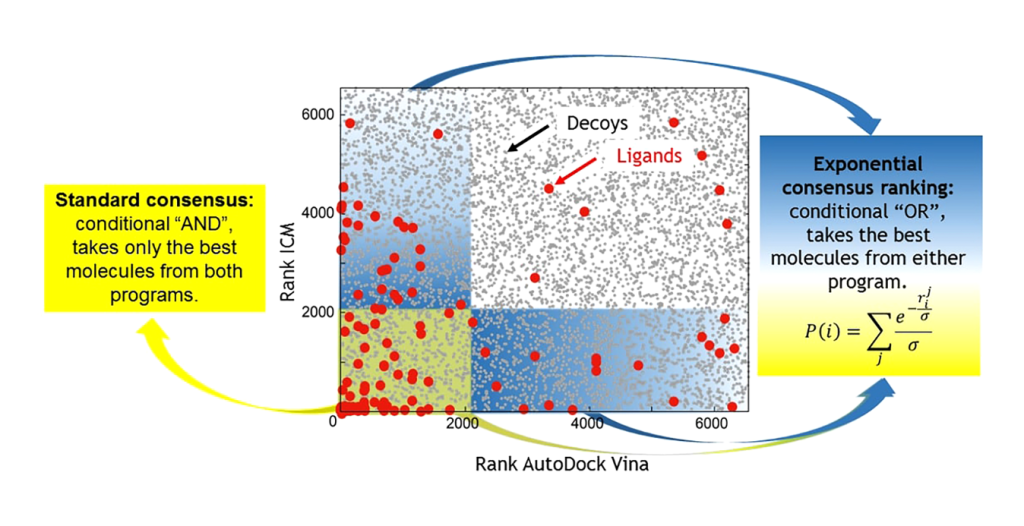
A variety of software tools are available for molecular docking, each offering different conformational search algorithms and scoring function combinations. Consensus docking, which involves using multiple scoring functions to validate docking results, is a recommended practice. While combining three or four scoring functions can enhance results, achieving sufficient predictive power remains challenging, especially for highly flexible ligands. Consequently, new approaches incorporating machine learning are emerging to improve scoring functions.
The Future of Drug Discovery
Molecular docking represents a vital tool in the modern pharmacological arsenal, offering a sophisticated method for identifying potential drug candidates. By leveraging advanced computational techniques and integrating machine learning, researchers are poised to make significant strides in the discovery and development of new drugs. As technology continues to evolve, the accuracy and efficiency of molecular docking will only improve, paving the way for more effective and personalized treatments in the future.
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Subscribe
to get our
LATEST NEWS




Related Posts

Molecular Biology & Biotechnology
Myosin’s Molecular Toggle: How Dimerization of the Globular Tail Domain Controls the Motor Function of Myo5a
Myo5a exists in either an inhibited, triangulated rest or an extended, motile activation, each conformation dictated by the interplay between the GTD and its surroundings.

Bioinformatics & Multiomics
Mapping the Invisible Arrows: Unraveling Disease Causality Through Network Biology
What began as a methodological proposition—constructing causality through three structured networks—has evolved into a vision for the future of systems medicine.


Read More Articles


Medicinal Chemistry & Pharmacology
April 15, 2025
Designing Better Sugar Stoppers: Engineering Selective α-Glucosidase Inhibitors via Fragment-Based Dynamic Chemistry
One of the most pressing challenges in anti-diabetic therapy is reducing the unpleasant and often debilitating gastrointestinal side effects that accompany α-amylase inhibition.





