In recent years, the realm of non-coding RNA (ncRNA) research has witnessed a paradigm shift, thanks to the emergence of sophisticated computational tools. These in silico methodologies have transformed the landscape of ncRNA target prediction, allowing researchers to delve deeper into the intricate mechanisms of gene regulation. By leveraging computational resources, scientists can now identify potential ncRNA candidates and their target genes with remarkable precision and efficiency.
The accessibility of various computational tools and databases plays a crucial role in this endeavor. Designed with user-friendliness in mind, these web-based platforms empower researchers without requiring extensive programming skills. This democratization of technology has broadened the scope of ncRNA research, enabling more scientists to participate in the discovery process. However, it is important to note that the predictive power of a single computational tool may be limited. Combining multiple approaches can significantly enhance the accuracy of identifying experimentally verifiable ncRNA-target interactions.
Predictive Tools for ncRNA Targeting
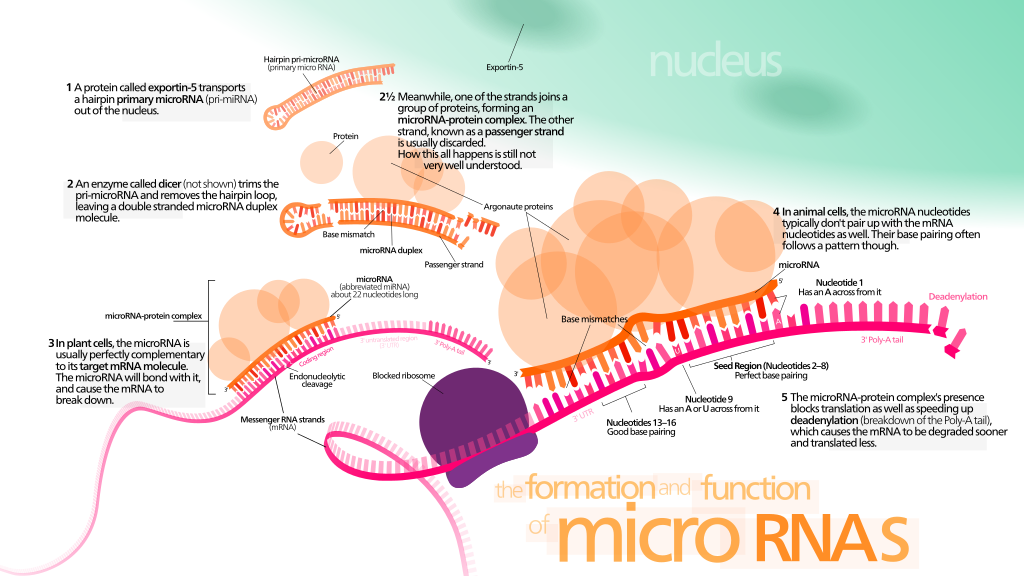
The landscape of ncRNA target prediction is rich with publicly available tools and databases. Among the most notable is miRBase, a comprehensive repository of miRNA sequences and annotations. With its vast collection of precursor and mature miRNA entries, miRBase provides an invaluable resource for researchers looking to explore miRNA interactions. These small RNA molecules repress gene expression by binding to miRNA response elements (MREs) on target RNAs, guided by the crucial seed region within the miRNA sequence.
Tools like DIANA-LncBase further extend the scope of ncRNA research by focusing on miRNA interactions with long non-coding RNAs (lncRNAs). This database curates a wealth of experimentally supported miRNA-lncRNA interactions, offering a robust platform for researchers investigating the regulatory roles of miRNAs beyond mRNAs.
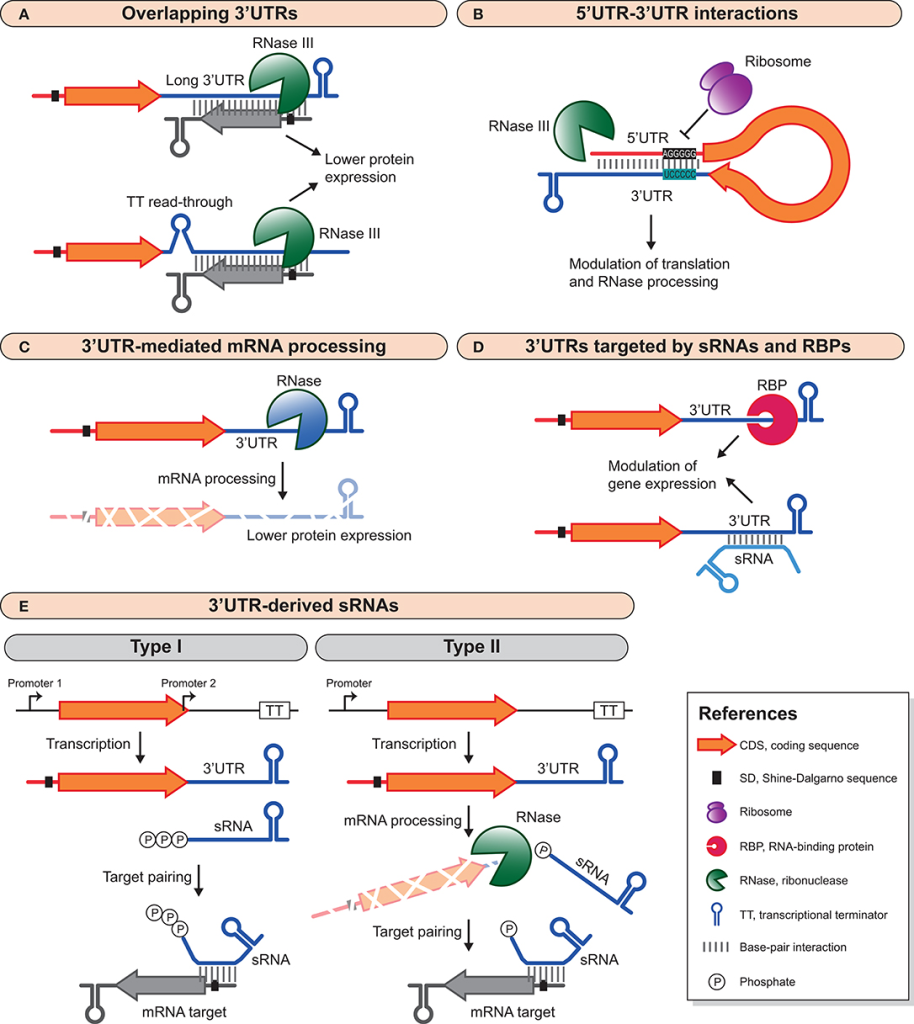
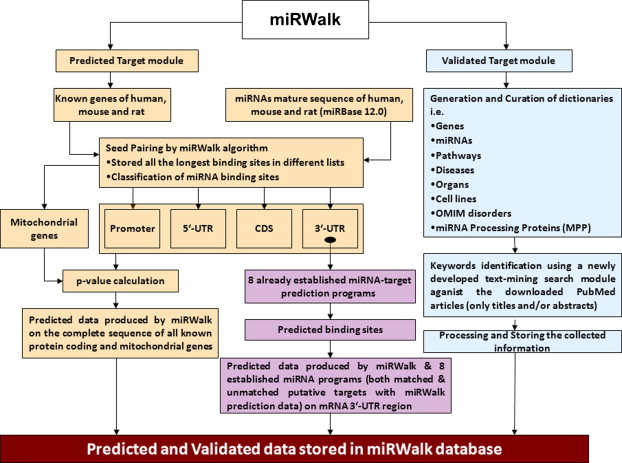
To navigate the diverse landscape of miRNA target prediction, researchers must consider several factors. False positives are a common challenge, as different algorithms may yield divergent results based on varying stringencies and predictive parameters. Tools like miRWalk address this issue by integrating predictions from multiple sources, enhancing the reliability of the identified interactions. This combinatorial approach is pivotal in improving the predictive confidence of ncRNA-target interactions.
Evaluating Binding Affinity and Site Accessibility
Understanding the biochemical features that underpin ncRNA-target interactions is essential for accurate predictions. The stability and efficiency of these interactions are often dictated by binding affinity and site accessibility. mRNAs, with their complex secondary structures, present both challenges and opportunities for ncRNA binding. Regions such as the 3′-untranslated regions (3′-UTRs) are typically more accessible and thus preferentially targeted by miRNAs.
The minimum free energy (MFE) of hybridization between ncRNAs and their targets serves as a key indicator of binding affinity. Lower MFE values suggest stronger, more likely interactions. Tools like RNAhybrid and miRWalk enable researchers to calculate these energies, providing a quantitative basis for predicting ncRNA-mRNA interactions. For instance, RNAhybrid allows users to input sequences and adjust parameters to identify high-affinity binding sites, thereby refining the list of potential interactions for experimental validation.
For larger RNA molecules, the ViennaRNA web services offer a suite of tools to analyze the structural and thermodynamic properties of RNA-RNA interactions. RNAfold and RNAcofold are particularly useful for predicting the secondary structures and binding affinities of lncRNAs and their targets. These tools not only enhance the understanding of ncRNA interactions but also facilitate the generation of publication-ready visualizations, making them accessible to both novice and experienced researchers.

Correlating ncRNA and mRNA Expression
In silico analysis extends beyond prediction to encompass the correlation of ncRNA and mRNA expression profiles. High-throughput technologies, such as microarrays and next-generation sequencing, have generated vast amounts of expression data. By mining these datasets, researchers can uncover the regulatory relationships between ncRNAs and their target mRNAs.
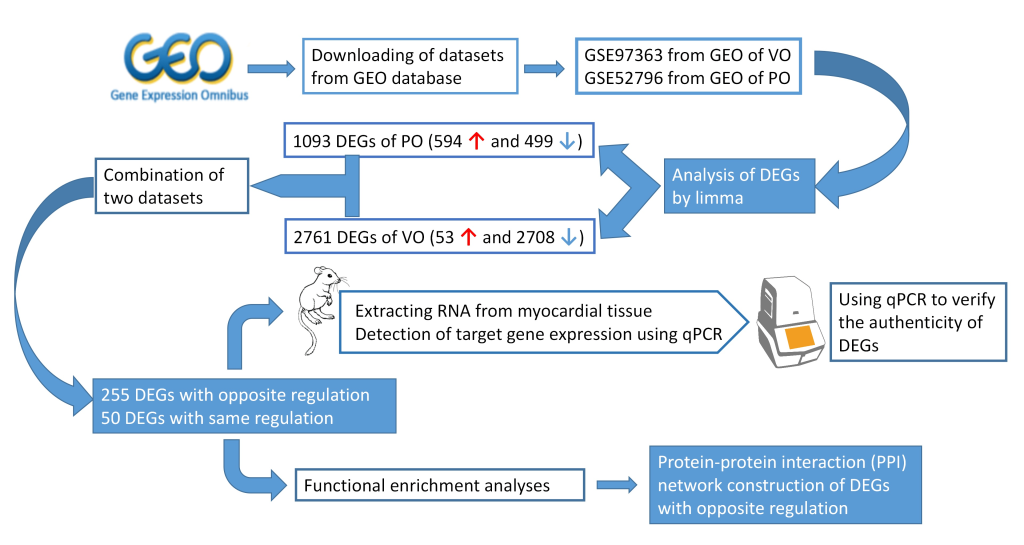
The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO) are two prominent repositories that provide extensive genomic data for such analyses. TCGA, for instance, offers paired miRNA and mRNA expression profiles from the same samples, enabling researchers to eliminate biases caused by inter-sample variation. This feature is particularly valuable in cancer research, where gene expression can vary widely between tumor and non-tumor tissues.

Similarly, GEO hosts a vast array of high-throughput genomic data across various species and experimental conditions. Researchers can leverage these resources to perform integrative analyses, revealing novel ncRNA-mRNA interaction networks and their roles in biological processes. Expression Atlas further enriches this landscape by providing comprehensive data on RNA and protein expression across different tissues and conditions, facilitating a deeper understanding of ncRNA functions.
Harnessing the Potential of ncRNA Research
The advent of in silico tools has revolutionized the field of ncRNA research, providing unprecedented insights into the complex regulatory networks that govern gene expression. By integrating computational predictions with experimental validation, researchers can unlock the full potential of ncRNAs as key regulators in various biological processes. As the repository of computational tools and databases continues to grow, so too does the promise of ncRNA research, paving the way for new discoveries and therapeutic innovations.
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Subscribe
to get our
LATEST NEWS




Related Posts

Bioinformatics & Multiomics
Mapping the Invisible Arrows: Unraveling Disease Causality Through Network Biology
What began as a methodological proposition—constructing causality through three structured networks—has evolved into a vision for the future of systems medicine.

Bioinformatics & Multiomics
Open-Source Bioinformatics: High-Resolution Analysis of Combinatorial Selection Dynamics
Combinatorial selection technologies are pivotal in molecular biology, facilitating biomolecule discovery through iterative enrichment and depletion.


Read More Articles

Drug Discovery Biology
April 17, 2025
Myosin’s Molecular Toggle: How Dimerization of the Globular Tail Domain Controls the Motor Function of Myo5a
Myo5a exists in either an inhibited, triangulated rest or an extended, motile activation, each conformation dictated by the interplay between the GTD and its surroundings.


Medicinal Chemistry & Pharmacology
April 15, 2025
Designing Better Sugar Stoppers: Engineering Selective α-Glucosidase Inhibitors via Fragment-Based Dynamic Chemistry
One of the most pressing challenges in anti-diabetic therapy is reducing the unpleasant and often debilitating gastrointestinal side effects that accompany α-amylase inhibition.





{kind=link}