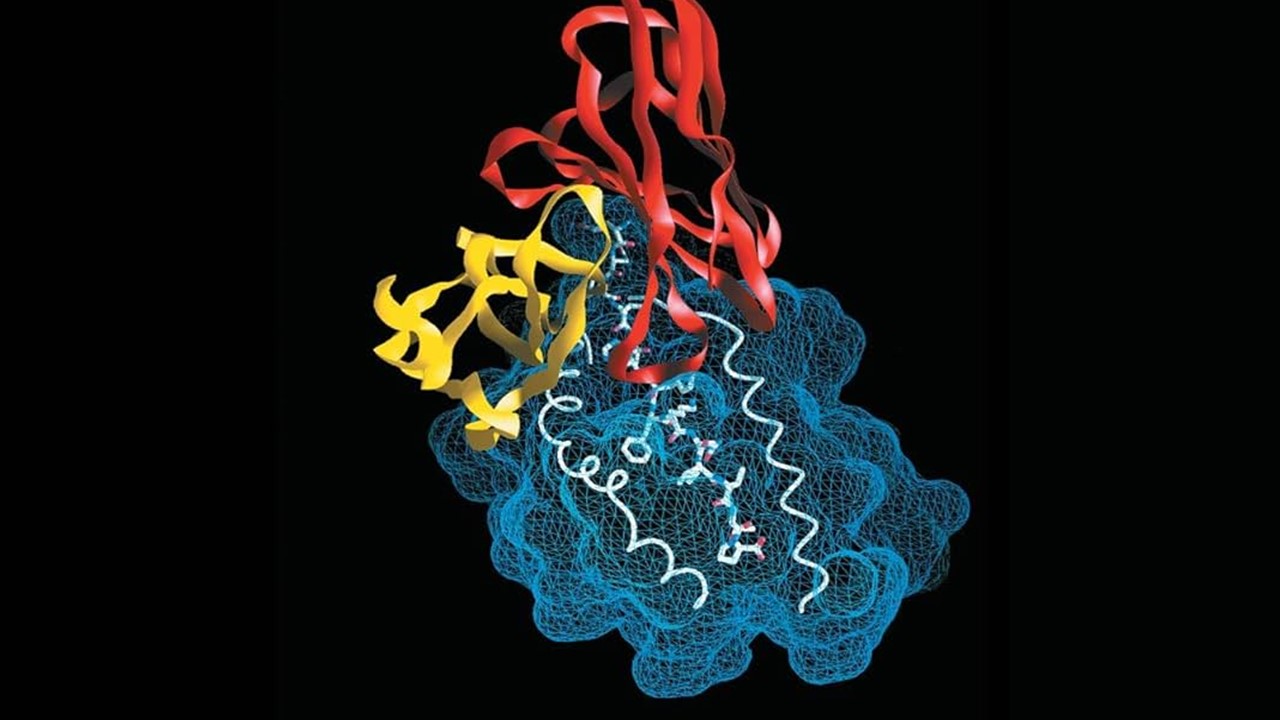
The Undruggable Era: A Legacy of Frustration
For decades, the term “undruggable” haunted drug discovery. It described proteins like KRAS, Myc, and p53—targets central to cancer, neurodegeneration, and immune disorders but structurally defiant. These proteins lacked the deep hydrophobic pockets that small molecules traditionally exploit. KRAS, for instance, cycles between GTP-bound active and GDP-bound inactive states, its surface as smooth as polished stone. Early attempts to target it, like farnesyltransferase inhibitors, failed spectacularly, cementing its reputation as a “biological dead end.”
The problem extended beyond RAS. Transcription factors like Myc, which drive 70% of cancers, flaunt disordered structures that evade small-molecule binding. p53, the “guardian of the genome,” is inactivated in half of all tumors but resists drug design due to its reliance on transient interactions with MDM2 for degradation. These targets became symbols of futility, their mechanisms deemed too dynamic, too featureless, or too integrated into cellular networks to attack.
Yet desperation bred innovation. The 2013 discovery of a cryptic pocket in KRASG12C—a mutant subtype common in lung cancer—ignited a revolution. Using covalent chemistry, researchers designed compounds that latched onto a previously hidden cysteine residue, locking KRAS in its inactive state. This breakthrough, culminating in the 2021 FDA approval of sotorasib, didn’t just target KRAS—it shattered the myth of the undruggable.
Covalent Chemistry: The Art of Molecular Ambush
Covalent inhibitors are the assassins of drug discovery. They exploit reactive amino acids like cysteine or lysine, anchoring themselves to targets with irreversible bonds. Sotorasib and adagrasib, the first KRASG12C inhibitors, epitomize this strategy. By targeting Cys12—a mutation-specific residue—these drugs trap KRAS in its GDP-bound form, starving tumors of the growth signals they crave.
The brilliance lies in their precision. Unlike early covalent drugs, which relied on brute-force reactivity, modern agents use “targeted covalent design.” Warheads like acrylamides are tuned to react only when positioned perfectly within a binding pocket. For example, osimertinib, a third-generation EGFR inhibitor, exploits Cys797 in the ATP-binding site to overcome resistance mutations in lung cancer. Its acrylamide group acts like a molecular latch, snapping shut only when the drug aligns with the mutant kinase.
Yet challenges persist. Off-target reactivity can trigger toxicity, as seen with ibrutinib’s unintended inhibition of ITK in immune cells. And resistance looms—KRASG12C mutants have already evolved secondary mutations like Y96D that weaken drug binding. To stay ahead, researchers are engineering “reversible covalent” inhibitors that form transient bonds, balancing potency with adaptability.
Allostery: The Puppeteers of Protein Conformation
If covalent drugs are assassins, allosteric modulators are puppeteers. They pull hidden strings, inducing conformational changes that disable targets from afar. Take SHP2, a phosphatase that funnels signals from growth factor receptors to RAS. Its role in cancers like NSCLC and leukemia made it a priority, but its active site—a shallow, charged cleft—defied conventional inhibition.
The solution? Target its “switch.” In its inactive state, SHP2 folds into a closed conformation, its N-SH2 domain blocking the catalytic site. Drugs like TNO155 stabilize this autoinhibited state, preventing SHP2 from adopting the open structure needed for signaling. This approach, validated by cryo-EM structures, has shown promise in early trials, shrinking tumors in RAS-driven cancers.
KRAS itself is now an allosteric playground. MRTX-1133, a compound in development for KRASG12D pancreatic cancer, binds a pocket near the protein’s switch II loop. Unlike covalent inhibitors, MRTX-1133 avoids competing with GTP, instead distorting the region where RAF kinases dock. This prevents downstream MAPK activation while sparing wild-type KRAS—a feat of molecular surgery.
Protein-Protein Interaction Inhibitors: Sabotaging Cellular Handshakes
Protein-protein interactions (PPIs) are the handshakes of biology—often fleeting, always critical. Targeting them requires disrupting interfaces spanning thousands of square angstroms, a task once deemed impossible. Venetoclax, a Bcl-2 inhibitor, proved it wasn’t. By mimicking the BH3 helix of pro-apoptotic proteins like BIM, venetoclax plugs into Bcl-2’s hydrophobic groove, unleashing apoptosis in leukemia cells.
The p53-MDM2 axis is another battleground. In healthy cells, MDM2 tags p53 for degradation, keeping its tumor-suppressing power in check. Cancers hijack this system, hyperactivating MDM2 to neutralize p53. Nutlin-3, an early MDM2 inhibitor, showed that small molecules could block this interaction, but poor pharmacokinetics limited its use. Enter AMG-232, a next-gen compound with picomolar affinity. By precisely positioning hydrophobic groups into MDM2’s p53-binding cleft, AMG-232 resurrects p53’s function, driving cancer cells into senescence or death.
Yet PPIs remain a thorny frontier. Many interfaces lack deep pockets, demanding creative solutions like stapled peptides. ALRN-6924, a stabilized α-helix that blocks both MDM2 and MDMX, is in trials for liposarcoma. Its rigid structure mimics p53’s transactivation domain, outcompeting the native protein for binding—a molecular decoy with clinical teeth.
Targeted Degradation: Obliteration as a Therapeutic Strategy
Why inhibit a protein when you can erase it? PROTACs (proteolysis-targeting chimeras) answer this question by hijacking the cell’s garbage disposal. These heterobifunctional molecules link a target-binding warhead to an E3 ligase recruiter, marking proteins for proteasomal destruction. ARV-471, an estrogen receptor (ER) degrader, exemplifies this approach. In hormone-resistant breast cancer, ARV-471 reduces ER levels by 90%, bypassing mutations that render traditional anti-estrogens useless.
KRASG12C degraders like LC-2 take this further. By fusing a covalent KRAS binder to a VHL ligase ligand, LC-2 eliminates the oncoprotein entirely, circumventing resistance mechanisms that plague inhibitors. Early data show tumor regressions in models resistant to sotorasib, suggesting degradation could outmaneuver evolution itself.
But PROTACs are just the beginning. LYTACs (lysosome-targeting chimeras) extend degradation to extracellular proteins like PD-L1 by hijacking cell-surface receptors. AUTACs (autophagy-targeting chimeras) traffic targets to autophagosomes, offering a path to degrade organelles like dysfunctional mitochondria. These platforms are rewriting oncology, neurology, and immunology—one protein at a time.
Nucleic Acid Therapeutics: Silencing the Source
If proteins are the actors, nucleic acids are the scriptwriters. Targeting DNA or RNA offers a way to rewrite the play entirely. AZD4785, an antisense oligonucleotide (ASO), silences KRAS by binding its mRNA, promoting degradation by RNase H. In preclinical models, AZD4785 shrinks lung tumors without harming normal tissues—a feat of genetic discrimination.
CRISPR-Cas9 takes this further. Researchers have engineered exosomes loaded with Cas9 and KRASG12D-targeting guide RNAs. In pancreatic cancer models, these nanovehicles selectively disrupt the oncogene, sparing wild-type cells. Meanwhile, base editors are being tested to correct KRASG12D to benign variants like G12V, offering a one-time fix for genetic havoc.
For undruggable transcription factors like Myc, G-quadruplex stabilizers offer a workaround. Quarfloxin, a fluoroquinolone derivative, binds GC-rich DNA sequences in the Myc promoter, freezing the DNA into a knot-like structure. This halts transcription, starving tumors of Myc’s pro-growth signals—a masterstroke of topological interference.
Immunotherapy: Turning the Body Against the Undruggable
The immune system is the ultimate hitman—if you can point it at the right target. mRNA vaccines like mRNA-5671 encode fragments of mutant KRAS (G12D, G12V, etc.), training T-cells to recognize and destroy tumors bearing these neoantigens. Early trials show promising immune activation in pancreatic and lung cancers.
T-cell receptor (TCR) therapies take this further. By engineering T-cells to express receptors specific for KRASG12V or G12D peptides presented on MHC-I, researchers have achieved remissions in metastatic cancers. These “living drugs” patrol the body, eliminating mutant cells with ruthless precision.
Even checkpoint inhibitors play a role. Sotorasib synergizes with anti-PD1 antibodies in KRASG12C lung cancer, combining direct RAS inhibition with immune activation. The result? Deeper, more durable responses—a testament to the power of polypharmacy.
Overcoming Resistance: The Evolutionary Arms Race
Cancer evolves, and so must our strategies. KRASG12C inhibitors like sotorasib initially shrink tumors, but resistance often emerges via secondary mutations (Y96D, R68M) or adaptive MAPK reactivation. The solution? Hit multiple nodes at once. Combining sotorasib with SOS1 inhibitors (e.g., BI-3406) blocks feedback loops, while adding SHP2 inhibitors (TNO155) prevents pathway rewiring.
PROTACs offer another edge. By degrading targets entirely, they eliminate the substrate for resistance mutations. KRASG12C degraders like LC-2 are active against Y96D mutants, as the protein itself is vaporized. Similarly, MDM2 degraders (e.g., MD-224) prevent tumors from upregulating alternative E3 ligases.
AI is accelerating this arms race. Tools like AlphaFold2 predict how mutations alter protein structure, guiding the design of next-gen inhibitors. Schrödinger’s FEP+ models drug-target interactions at quantum mechanical resolution, optimizing compounds before synthesis begins.
The Future: Beyond the Undruggable Horizon
The undruggable era is ending. MYC, long considered untouchable, is being targeted via WDR5 inhibitors that disrupt its partnership with chromatin. OMO-103, a stapled peptide in trials, blocks Myc-Max dimerization, choking tumors of their growth signals.
In neurodegeneration, Tau aggregates—once impervious to drugs—are being cleared with AUTOTACs. These degraders exploit the proteasome’s unfoldase activity, dismantling the fibrils that strangle neurons in Alzheimer’s.
The next frontier? Time-resolved pharmacology. Drugs that activate only in hypoxic tumor microenvironments or release payloads in response to specific enzymes. Imagine a covalent KRAS inhibitor inert in normal tissues but reactive in acidic, ROS-rich tumors—a therapeutic smart bomb.
The New Druggable
The conquest of undruggable targets is more than a technical triumph—it’s a paradigm shift. We’ve moved from viewing proteins as static targets to dynamic entities with exploitable weaknesses. Covalent chemistry, allostery, degradation, and immunotherapy are not just tools but lenses, revealing vulnerabilities invisible to past generations.
As cryo-EM maps once-hidden pockets, AI predicts resistance mutations before they arise, and CRISPR edits disease at its roots, the line between “druggable” and “undruggable” blurs. What remains is a challenge not of possibility, but of imagination. The proteins that once mocked our efforts now stand as monuments to human ingenuity—proof that in science, the impossible is always temporary.
Study DOI: https://doi.org/10.1038/s41392-023-01589-z
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Editor-in-Chief, PharmaFEATURES

Subscribe
to get our
LATEST NEWS




Related Posts

Medicinal Chemistry & Pharmacology
Aerogel Pharmaceutics Reimagined: How Chitosan-Based Aerogels and Hybrid Computational Models Are Reshaping Nasal Drug Delivery Systems
Simulating with precision and formulating with insight, the future of pharmacology becomes not just predictive but programmable, one cell at a time.
Medicinal Chemistry & Pharmacology
Coprocessed for Compression: Reengineering Metformin Hydrochloride with Hydroxypropyl Cellulose via Coprecipitation for Direct Compression Enhancement
In manufacturing, minimizing granulation lines, drying tunnels, and multiple milling stages reduces equipment costs, process footprint, and energy consumption.

Medicinal Chemistry & Pharmacology
Decoding Molecular Libraries: Error-Resilient Sequencing Analysis and Multidimensional Pattern Recognition
tagFinder exemplifies the convergence of computational innovation and chemical biology, offering a robust framework to navigate the complexities of DNA-encoded science