In the realm of pharmaceuticals, ensuring the safety and efficacy of drugs is paramount. The journey from the laboratory bench to the medicine cabinet of patients is an intricate process fraught with challenges and ethical considerations. This comprehensive article delves into the crucial role played by the Food and Drug Administration (FDA) in safeguarding public health and the meticulous conduct of clinical trials that underpin drug approvals. We explore the historical backdrop of regulatory evolution, the ethical principles governing clinical trials, and the complex interplay of factors determining a drug’s safety and effectiveness.
Role of the FDA: Safeguarding Public Health
Historical Evolution of Drug Regulation
The FDA, a pivotal federal regulatory agency within the U.S. Department of Health and Human Services, is entrusted with a multifaceted mission: protecting the health of the public by ensuring the safety, efficacy, and security of an array of products, including drugs, biological products, medical devices, food, cosmetics, and radiation-emitting products. The FDA’s role extends beyond safeguarding; it actively contributes to enhancing public health by facilitating innovations that make medicines and foods more effective, safer, and more affordable. Additionally, it equips individuals with science-based information to enhance their health outcomes.

The inception of drug regulation in the United States can be traced back to the Federal Food and Drug Act of 1906, which primarily focused on the interstate transport of adulterated or misbranded foods and drugs. Astonishingly, this early legislation did not mandate the establishment of drug efficacy or safety standards. A turning point in drug regulation emerged in 1938 after a tragedy involving “elixir sulfanilamide” claimed the lives of over 100 children. This incident prompted the enforcement of amended legislation entrusted to the FDA, necessitating toxicity studies and the submission of New Drug Applications (NDAs) for drug promotion and distribution. Remarkably, while safety data became mandatory, efficacy demonstration was not yet required.

The Thalidomide Crisis and the Harris-Kefauver Amendments
The 1960s witnessed a global crisis with the introduction of thalidomide, a hypnotic drug lacking apparent advantages over existing alternatives. Extensive epidemiological research revealed that thalidomide, when taken early in pregnancy, triggered a severe and rare birth defect called phocomelia. In response to this devastating event, the U.S. Congress passed the Harris-Kefauver amendments to the Food, Drug, and Cosmetic Act in 1962. These pivotal amendments ushered in the era of rigorous proof of efficacy alongside comprehensive documentation of relative safety, especially concerning the risk-to-benefit ratio tailored to the treated disease’s gravity. This marked a significant leap in regulatory standards and was pivotal in reshaping the landscape of drug development.

Contemporary Challenges for the FDA
In contemporary times, the FDA confronts a formidable challenge, particularly in light of resource constraints imposed by Congress. The common perception is that the agency’s mission may be unattainable with limited resources. Furthermore, the imperfections within the approval process not only pose risks of unanticipated adverse effects but also lead to delays in bringing drugs with significant therapeutic benefits to the market. These challenges underscore the intricate balance between safety and timely access to potentially life-saving therapies.
The Conduct of Clinical Trials: A Rigorous Path to Approval
Ethical Principles Governing Clinical Trials
Clinical trials represent the crucible where the safety, efficacy, and pharmacokinetic properties of candidate drugs in humans are meticulously scrutinized. The National Institutes of Health (NIH) articulate seven ethical principles that must be satisfied before a clinical trial commences. These principles encompass social and clinical value, scientific validity, fair subject selection, informed consent, a favorable risk-benefit ratio, independent review, and respect for potential and enrolled subjects.
Phases of Clinical Trials
FDA-regulated clinical trials typically unfold in four distinct phases. Phases I through III are meticulously designed to establish the safety and efficacy of the investigational drug, while phase IV postmarketing trials delve into additional information regarding new indications, risks, and optimal dosing regimens. Each phase necessitates an exhaustive evaluation, culminating in the submission of an NDA or Biologics License Application (BLA) to the FDA.
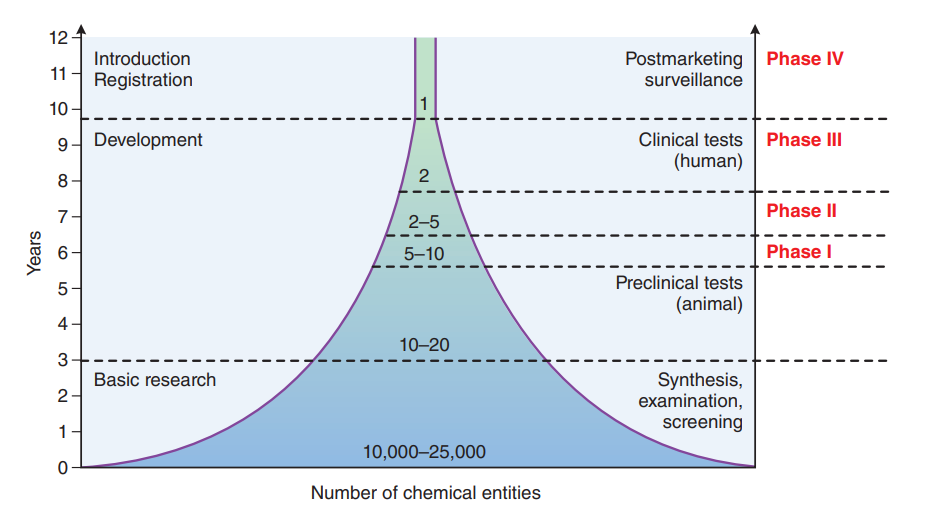
The Role of PDUFA
The Prescription Drug User Fee Act (PDUFA), enacted in 1992 and subsequently renewed, heralded a pivotal paradigm shift in drug development. Under PDUFA, pharmaceutical companies significantly contribute to the FDA’s budget through user fees, aimed at expediting drug approval reviews by augmenting resources. This legislative initiative not only enhances drug safety but also amplifies resources for reviewing television drug advertising. The collaborative approach between the FDA and the pharmaceutical industry has streamlined the drug approval timeline, with reviews typically spanning 6 to 10 months after an NDA submission.
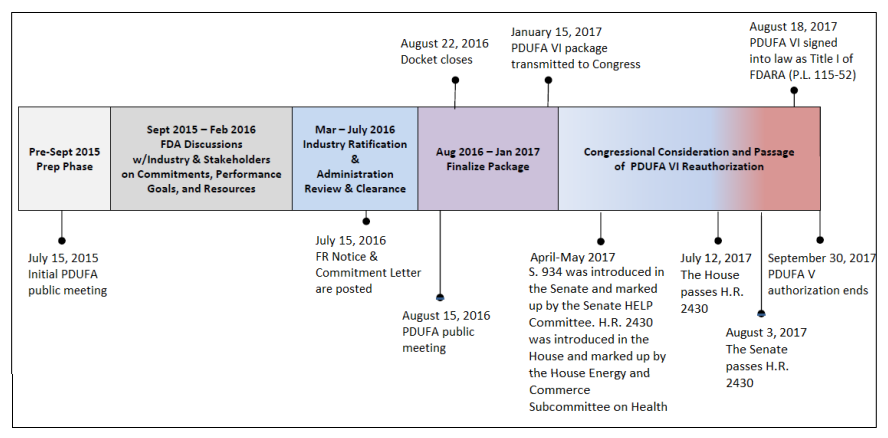
UserFees/PrescriptionDrugUserFee/UCM516686.pdf. CRS added the April through August 2017 text and
modified other text in the figure.
The Crucial Role of Labeling
Before a drug can be marketed, the FDA and the pharmaceutical company must concur on the content of the official prescribing information, commonly known as the “label.” This label encompasses approved indications, clinical pharmacological data, dosage guidelines, adverse reactions, and special warnings and precautions. It serves as a pivotal reference for healthcare professionals, guiding safe and effective drug use.

In addition to its pivotal role in drug approval, the U.S. Food and Drug Administration (FDA) serves as the primary regulatory authority responsible for ensuring the safety, efficacy, and security of a diverse range of products essential to public health. This multifaceted agency also oversees the approval and regulation of food products, dietary supplements, medical devices, and cosmetics, collectively safeguarding the American populace by upholding rigorous standards and conducting thorough evaluations across various industries critical to consumer well-being.

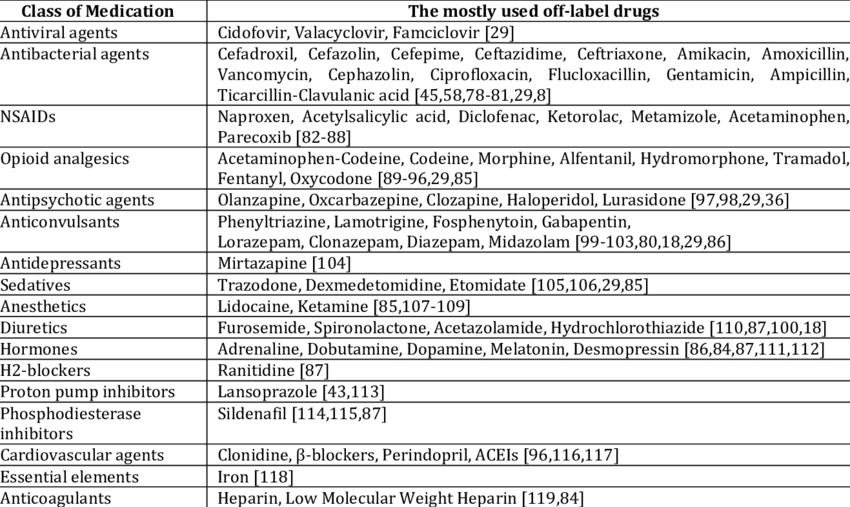
Off-Label Prescribing and Its Implications
While physicians in the United States possess the legal authority to prescribe drugs for off-label indications based on their clinical judgment, third-party payers typically withhold reimbursement unless the new use aligns with a statutorily named compendium. This practice aims to strike a balance between clinical autonomy and ensuring patient safety. Deviating from approved indications can expose physicians to potential litigation if adverse effects arise from unapproved drug uses.


Determining “Safe” and “Effective”
The Complex Landscape of Efficacy Evaluation
Establishing efficacy for FDA approval entails conducting “adequate and well-controlled investigations,” usually involving two replicate clinical trials. These trials are often randomized, double-blind, and placebo-controlled, but exceptions exist. The choice of a placebo control becomes contentious when alternative treatments are available, aligning with the World Medical Association’s Declaration of Helsinki, which discourages placebo controls when an effective treatment option exists.
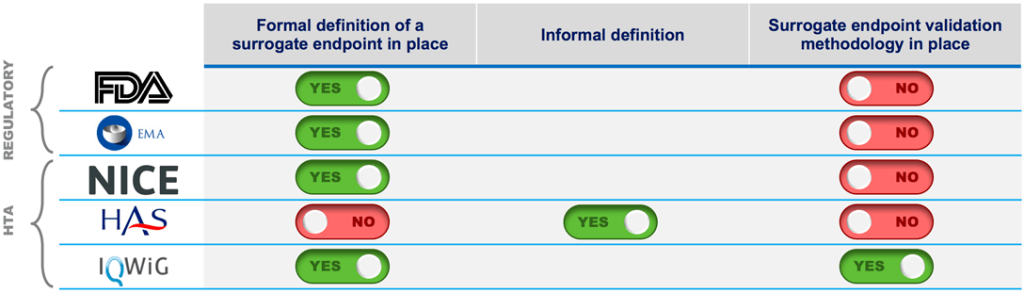
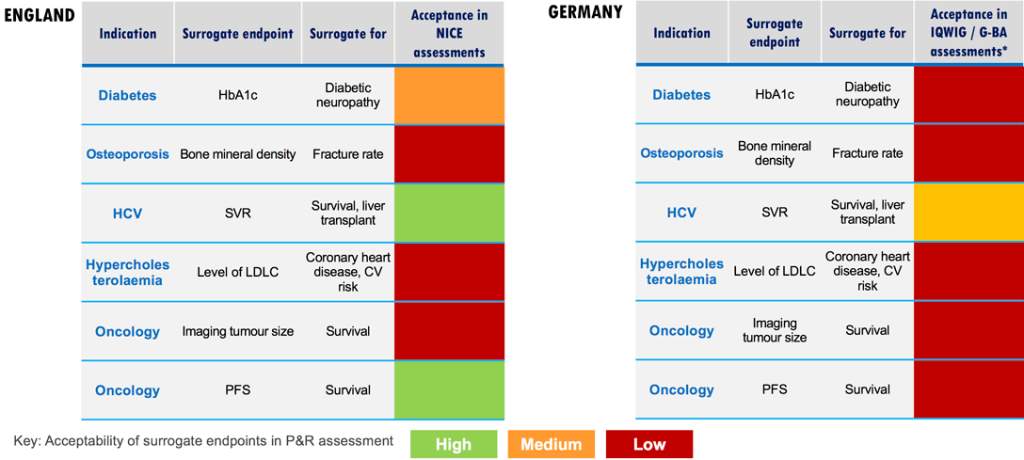
Surrogate Endpoints and Their Challenges
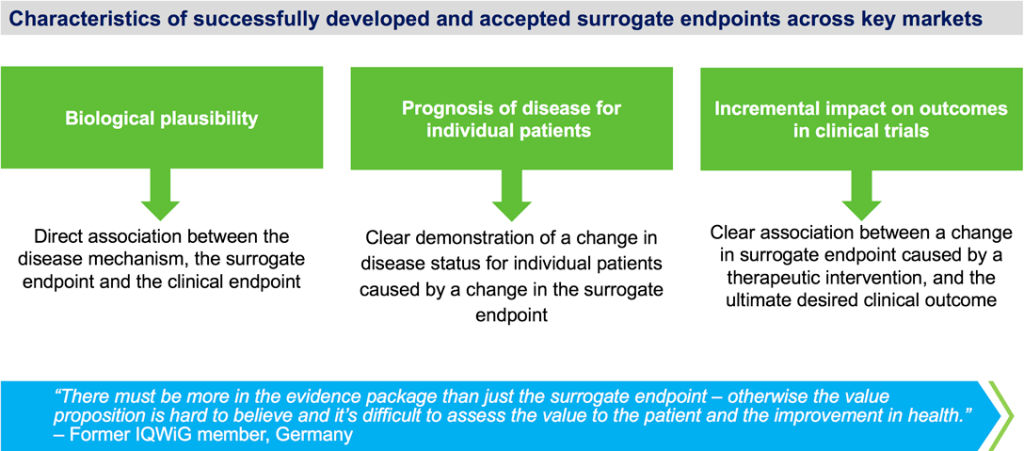
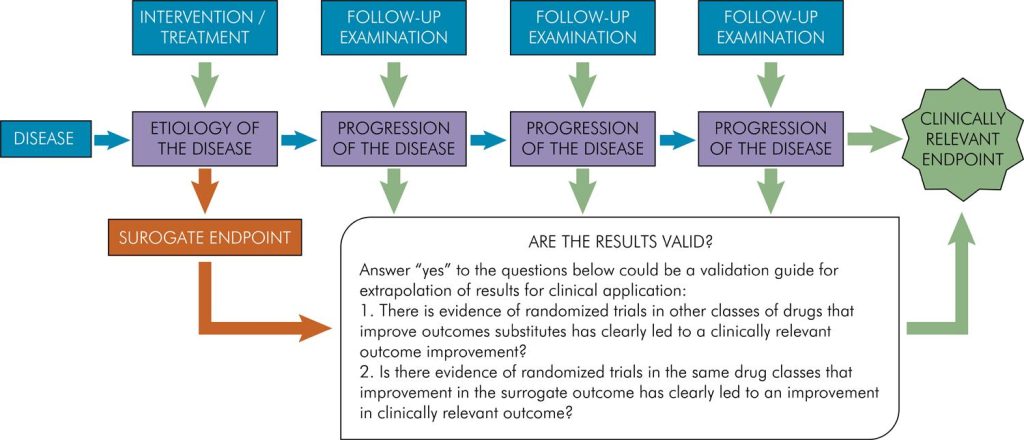
Clinical trials often rely on surrogate endpoints, quantifiable parameters predictive of relevant clinical outcomes. Examples include LDL cholesterol as a predictor of myocardial infarction or bone mineral density as a predictor of fractures. While surrogate endpoints expedite trials, their relevance to the treated disease remains a subject of scrutiny. The example of ezetimibe, an LDL-lowering drug, illustrates the complexities. Approval based on LDL reduction was later questioned when clinical trials failed to demonstrate direct clinical benefits. Balancing efficiency and clinical relevance remains an ongoing challenge.
Exploring Drug Safety
No drug is entirely devoid of adverse effects, with some occurring rarely and unpredictably. Standard phase III clinical trials, involving relatively small populations, may fail to detect these infrequent adverse events. Consequently, unveiling the true spectrum and incidence of unwanted effects often becomes apparent only during phase IV postmarketing surveillance. This stage, characterized by broader drug use, illuminates potential risks that could not be anticipated earlier.

Postmarketing Surveillance Strategies
Multiple strategies exist to detect adverse reactions after drug marketing. These include cohort studies, case-control studies, meta-analyses, and voluntary reporting systems. Voluntary reporting, involving healthcare professionals, third-party payers, consumers, and pharmacy committees, plays a pivotal role in generating early signals of adverse drug reactions. Notably, MedWatch, a 20-year-old reporting system in the U.S., continues to evolve, aiming to foster consumer engagement and reporting.
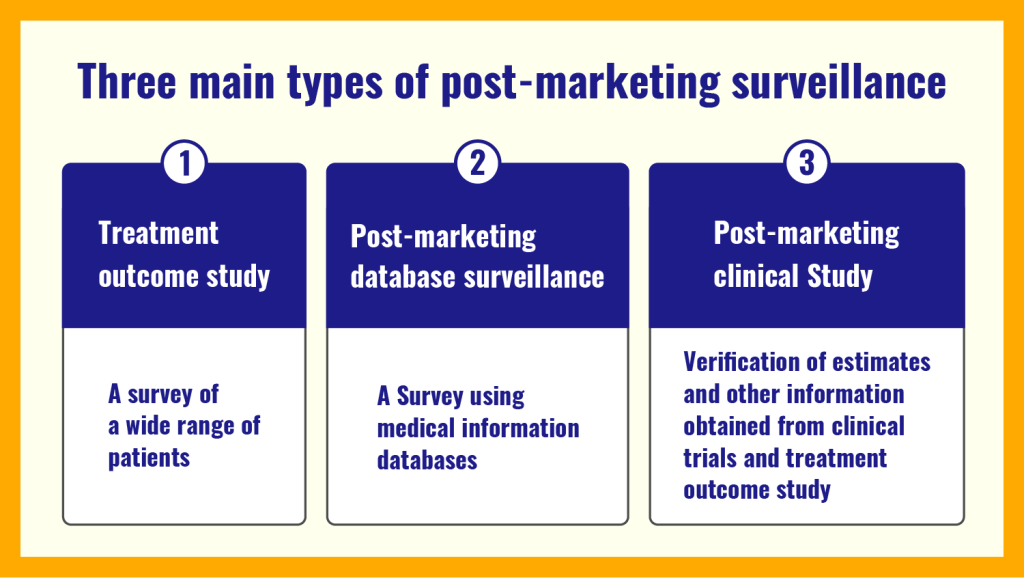

Post-marketing database investigation. Medical Data Vision Co., Ltd. (2023). What is Post-Marketing Surveillance (PMS)? A complete description of the 3 different types of PMS, how it differentiates from early-phase pharmaco-vigilance investigation (EPPV), and the overall flow of data. Retrieved from https://en.mdv.co.jp/ebm/column/article/29.html
 Difference between post-marketing surveillance and immediate post-marketing surveillance. Medical Data Vision Co., Ltd. (2023). What is Post-Marketing Surveillance (PMS)? A complete description of the 3 different types of PMS, how it differentiates from early-phase pharmaco-vigilance investigation (EPPV), and the overall flow of data. Retrieved from https://en.mdv.co.jp/ebm/column/article/29.html
Difference between post-marketing surveillance and immediate post-marketing surveillance. Medical Data Vision Co., Ltd. (2023). What is Post-Marketing Surveillance (PMS)? A complete description of the 3 different types of PMS, how it differentiates from early-phase pharmaco-vigilance investigation (EPPV), and the overall flow of data. Retrieved from https://en.mdv.co.jp/ebm/column/article/29.htmlIn conclusion, the intricate web of drug regulation, clinical trials, and postmarketing surveillance serves as a critical framework for ensuring the safety and efficacy of pharmaceuticals. The FDA, guided by evolving legislation and principles, plays a central role in this process. However, the quest for a delicate balance between safety and timely access to innovative therapies persists, underscoring the ongoing challenges in drug development and regulation. Ultimately, this pursuit stands as a testament to our commitment to advancing public health through science, ethics, and unwavering dedication.
Engr. Dex Marco Tiu Guibelondo, B.Sc. Pharm, R.Ph., B.Sc. CpE
Subscribe
to get our
LATEST NEWS




Related Posts
Regulatory Affairs
Decoding Drug Prices: Unraveling the Intricate Puzzle of Pharmaceutical Pricing
Only by untangling the web of pharmaceutical pricing can we pave the way for a more affordable and equitable healthcare system.
Regulatory Affairs
UK’s MHRA Authorizes Market Release of Eli Lilly’s Mirikizumab
Mirikizumab, Lilly’s breakthrough treatment for ulcerative colitis.


Read More Articles







Drug Discovery Biology
April 17, 2025
Myosin’s Molecular Toggle: How Dimerization of the Globular Tail Domain Controls the Motor Function of Myo5a
Myo5a exists in either an inhibited, triangulated rest or an extended, motile activation, each conformation dictated by the interplay between the GTD and its surroundings.
